MLCV@LCLS
Welcome to the Machine Learning and Computer Vision group (MLCV) in the Data Systems Division of the Linac Coherent Light Source (LCLS) at SLAC National Accelerator Laboratory!
We are a group of researchers and engineers working on developing and applying machine learning and computer vision techniques to the analysis of X-ray free-electron laser (XFEL) data. Our mission is to enable the next generation of XFEL experiments by providing cutting-edge data analysis tools and techniques. We bring innovative solutions from our partners in academia and industry to the forefront of XFEL science.
Do not hesitate to contact us if you are interested in joining our group or collaborating on a project!
news
| Dec 02, 2025 | Building a General Inference Engine for Molecular Structures and Ensembles Workshop - videos are online! |
|---|---|
| Oct 06, 2025 |
Enjoying nearby wildnerness after a great start to the Fall quarter. Wonderful moonrise to celebrate the mid Autumn Festival!

|
| Apr 18, 2025 |
Congratulations Axel for a stellar thesis defense! It has been a fun journey following you through your PhD, discovering new and better ways to look at life’s molecules! We’re excited to continue following your adventures in Boston with Philippine and Nausicaa :)

|
| Mar 26, 2025 |
We had a great time at the West Coast Structural Biology Workshop 2025! Co-organized by Mike Thompson (UC Merced), Alec Follmer (UC Davis)and Sandra Mous (LCLS), the workshop was a great success with more than 100 early-career participants.

|
| Mar 10, 2025 |
To wrap up the Winter quarter, we had a Journal Club on Statistical Crystallography, where Fred presented the paper “Statistical crystallography reveals an allosteric network in SARS-CoV-2 Mpro” by TJ Lane and colleagues. It was a great opportunity to learn about allostery and the nascent field of statistical crystallography and to discuss the implications for what we do at LCLS.

|
| Jan 01, 2025 | The MLCV website is now live! |
| Dec 15, 2024 | Our first Users from Latin America go home with at least 3 structures |
| Dec 07, 2024 | Building a General Inference Engine for Chemical Dynamics Workshop - videos are online! |
| Nov 21, 2024 | Daisy, Jacob, and Martin present their work at ICME 2024 |
| Nov 21, 2024 | John presents ARAMS at SC24 and ICME20 |
| Sep 27, 2024 | Jay wins the Outstanding Poster Award at the 2024 SSRL/LCLS Annual User's Meeting! |
| Aug 06, 2024 | Kevin gives the inaugural MLCV Seminar |
| Apr 01, 2024 | David opens his lab at Cold Spring Harbor Laboratory! |
| Sep 27, 2023 | Doris wins the Outstanding Poster Award at the 2023 SSRL/LCLS Annual User's Meeting! |
| Apr 06, 2023 | Axel delivers his SLAC Public Lecture! |
| Nov 16, 2022 | Fred presents at IPAM workshop on cryoEM |
selected publications
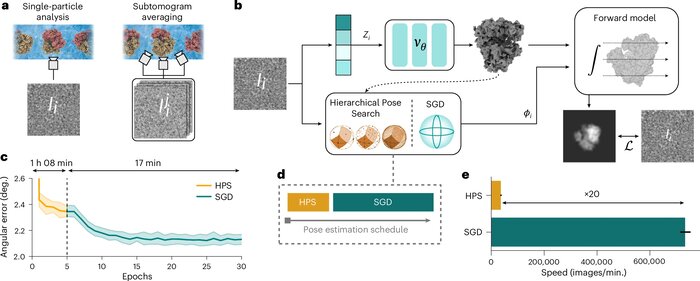
CryoDRGN-AI: neural ab initio reconstruction of challenging cryo-EM and cryo-ET datasets
Nature Methods (2025)
Proteins and other biomolecules form dynamic macromolecular machines that are tightly orchestrated to move, bind and perform chemistry. Cryo-electron microscopy and cryo-electron tomography can access the intrinsic heterogeneity of these complexes and are therefore key tools for understanding their function. However, three-dimensional reconstruction of the collected imaging data presents a challenging computational problem, especially without any starting information, a setting termed ab initio reconstruction. Here we introduce cryoDRGN-AI, a method leveraging an expressive neural representation and combining an exhaustive search strategy with gradient-based optimization to process challenging heterogeneous datasets. Using cryoDRGN-AI, we reveal new conformational states in large datasets, reconstruct previously unresolved motions from unfiltered datasets and demonstrate ab initio reconstruction of biomolecular complexes from in situ data. With this expressive and scalable model for structure determination, we hope to unlock the full potential of cryo-electron microscopy and cryo-electron tomography as a high-throughput tool for structural biology and discovery.
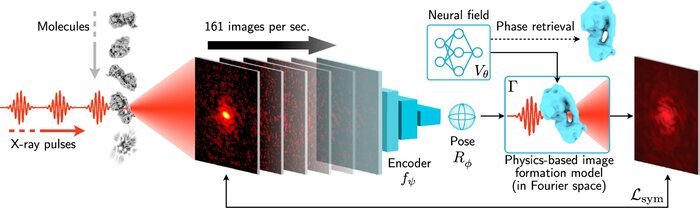
Scalable 3D reconstruction for X-ray single particle imaging with online machine learning
Nature Communications (2025)
X-ray free-electron lasers offer unique capabilities for measuring the structure and dynamics of biomolecules, helping us understand the basic building blocks of life. Notably, high-repetition-rate free-electron lasers enable single particle imaging, where individual, weakly scattering biomolecules are imaged under near-physiological conditions with the opportunity to access fleeting states that cannot be captured in cryogenic or crystallized conditions. Existing X-ray single particle reconstruction algorithms, which estimate the particle orientation for each image independently, are slow and memory-intensive when handling the massive datasets generated by emerging free-electron lasers. Here, we introduce X-RAI (X-Ray single particle imaging with Amortized Inference), an online reconstruction framework that estimates the structure of 3D macromolecules from large X-ray single particle datasets. X-RAI consists of a convolutional encoder, which amortizes pose estimation over large datasets, as well as a physics-based decoder, which employs an implicit neural representation to enable high-quality 3D reconstruction in an end-to-end, self-supervised manner. We demonstrate that X-RAI achieves state-of-the-art performance for small-scale datasets in simulation and challenging experimental settings and demonstrate its unprecedented ability to process large datasets containing millions of diffraction images in an online fashion. These abilities signify a paradigm shift in X-ray single particle imaging towards real-time reconstruction.
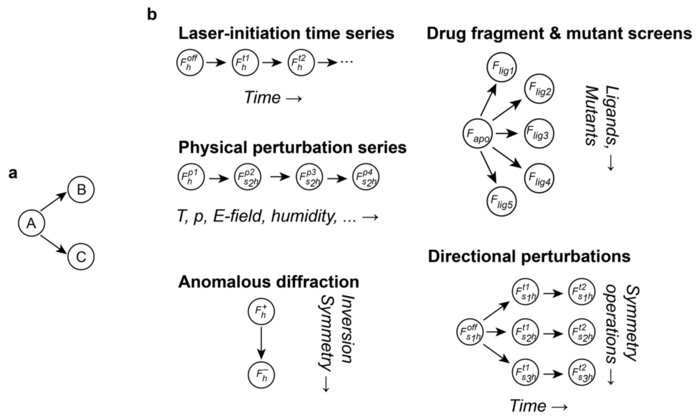
Sensitive Detection of Structural Differences using a Statistical Framework for Comparative Crystallography
bioRxiv (2024)
Chemical and conformational changes underlie the functional cycles of proteins. Comparative crystallography can reveal these changes over time, over ligands, and over chemical and physical perturbations in atomic detail. A key difficulty, however, is that the resulting observations must be placed on the same scale by correcting for experimental factors. We recently introduced a Bayesian framework for correcting (scaling) X-ray diffraction data by combining deep learning with statistical priors informed by crystallographic theory. To scale comparative crystallography data, we here combine this framework with a multivariate statistical theory of comparative crystallography. By doing so, we find strong improvements in the detection of protein dynamics, element-specific anomalous signal, and the binding of drug fragments.
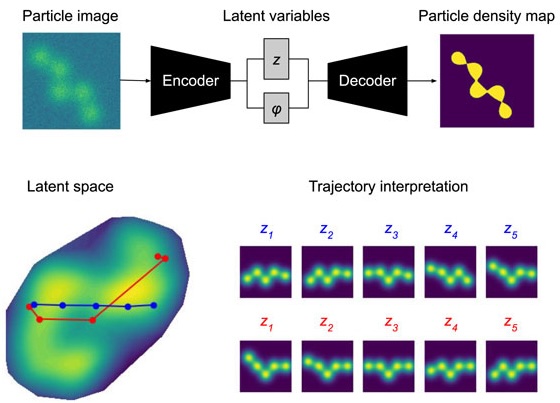
Towards interpretable Cryo-EM: disentangling latent spaces of molecular conformations
Frontiers in Molecular Biosciences (2024)
Molecules are essential building blocks of life and their different conformations (i.e., shapes) crucially determine the functional role that they play in living organisms. Cryogenic Electron Microscopy (cryo-EM) allows for acquisition of large image datasets of individual molecules. Recent advances in computational cryo-EM have made it possible to learn latent variable models of conformation landscapes. However, interpreting these latent spaces remains a challenge as their individual dimensions are often arbitrary. The key message of our work is that this interpretation challenge can be viewed as an Independent Component Analysis (ICA) problem where we seek models that have the property of identifiability. That means, they have an essentially unique solution, representing a conformational latent space that separates the different degrees of freedom a molecule is equipped with in nature. Thus, we aim to advance the computational field of cryo-EM beyond visualizations as we connect it with the theoretical framework of (nonlinear) ICA and discuss the need for identifiable models, improved metrics, and benchmarks. Moving forward, we propose future directions for enhancing the disentanglement of latent spaces in cryo-EM, refining evaluation metrics and exploring techniques that leverage physics-based decoders of biomolecular systems. Moreover, we discuss how future technological developments in time-resolved single particle imaging may enable the application of nonlinear ICA models that can discover the true conformation changes of molecules in nature. The pursuit of interpretable conformational latent spaces will empower researchers to unravel complex biological processes and facilitate targeted interventions. This has significant implications for drug discovery and structural biology more broadly. More generally, latent variable models are deployed widely across many scientific disciplines. Thus, the argument we present in this work has much broader applications in AI for science if we want to move from impressive nonlinear neural network models to mathematically grounded methods that can help us learn something new about nature.
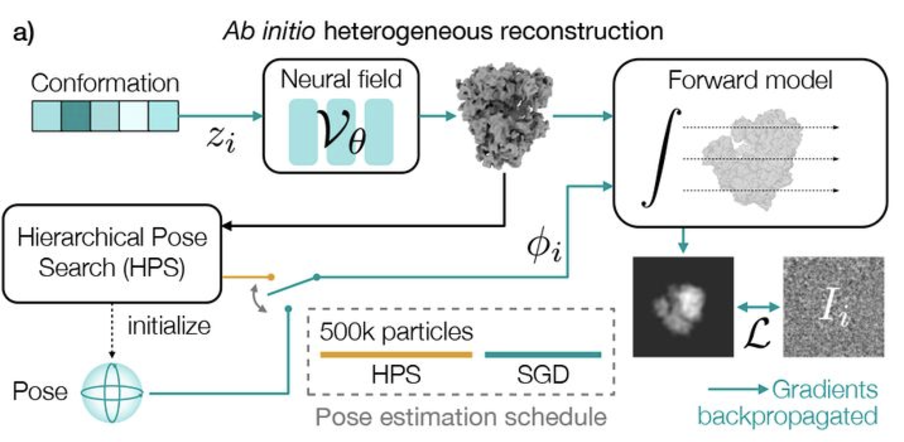
Revealing biomolecular structure and motion with neural ab initio cryo-EM reconstruction
bioRxiv (2024)
Proteins and other biomolecules form dynamic macromolecular machines that are tightly orchestrated to move, bind, and perform chemistry. Cryo-electron microscopy (cryo-EM) can access the intrinsic heterogeneity of these complexes and is therefore a key tool for understanding mechanism and function. However, 3D reconstruction of the resulting imaging data presents a challenging computational problem, especially without any starting information, a setting termed ab initio reconstruction. Here, we introduce a method, DRGN-AI, for ab initio heterogeneous cryo-EM reconstruction. With a two-step hybrid approach combining search and gradient-based optimization, DRGN-AI can reconstruct dynamic protein complexes from scratch without input poses or initial models. Using DRGN-AI, we reconstruct the compositional and conformational variability contained in a variety of benchmark datasets, process an unfiltered dataset of the DSL1/SNARE complex fully ab initio, and reveal a new “supercomplex” state of the human erythrocyte ankyrin-1 complex. With this expressive and scalable model for structure determination, we hope to unlock the full potential of cryo-EM as a high-throughput tool for structural biology and discovery.
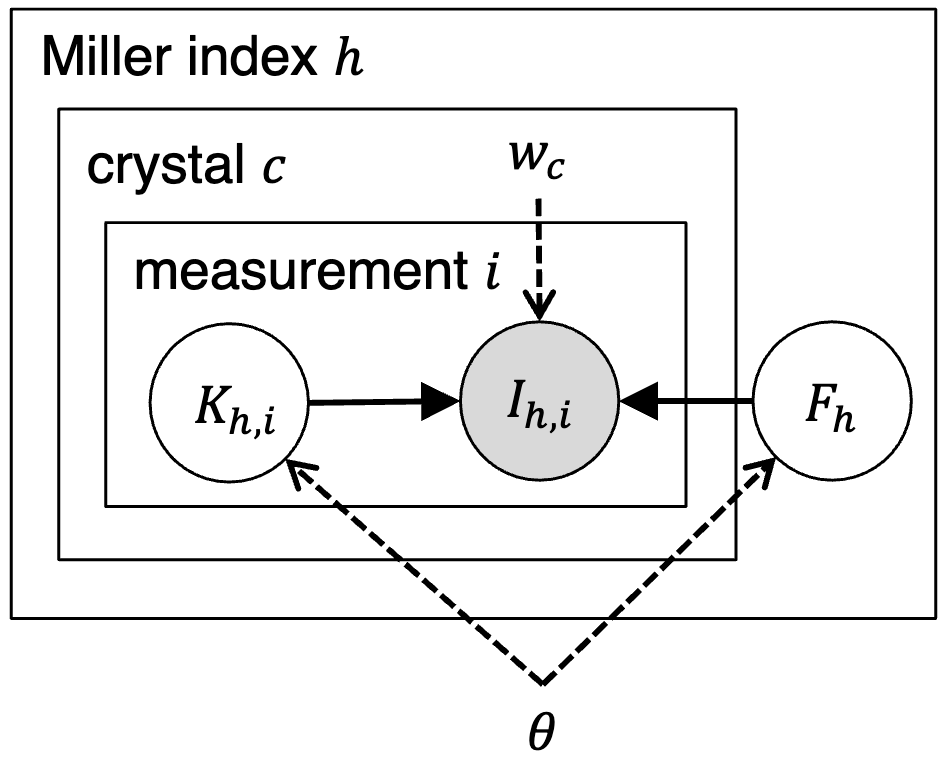
Assessing the applicability of Bayesian inference for merging small molecule microED data
ChemRxiv (2024)
Microcrystal electron diffraction (MicroED) is an emerging technique for characterizing small molecule structures from nanoscale crystals. Merging data from multiple crystals is a particularly challenging step in the microED workflow. A common practice is to manually curate datasets and apply scaling programs conventionally utilized in rotational X-ray diffraction (XRD), but this could be time-consuming and risks introducing human bias in data analysis. Recently, a Bayesian inference program named Careless (Dalton et al., 2022) has demonstrated excellent performance in merging macromolecular XRD data. Here, the applicability of Careless to small molecule microED data is evaluated and an investigation of the impact of dataset curation is performed. Benchmarking against XDS/XSCALE shows that Careless is an effective complementary approach that merges data to a higher CC1/2 value at high resolution. Furthermore, merging outcomes are not significantly improved by curating datasets either manually or with an automated extension to Careless, cautioning against the common practice of manual dataset curation.
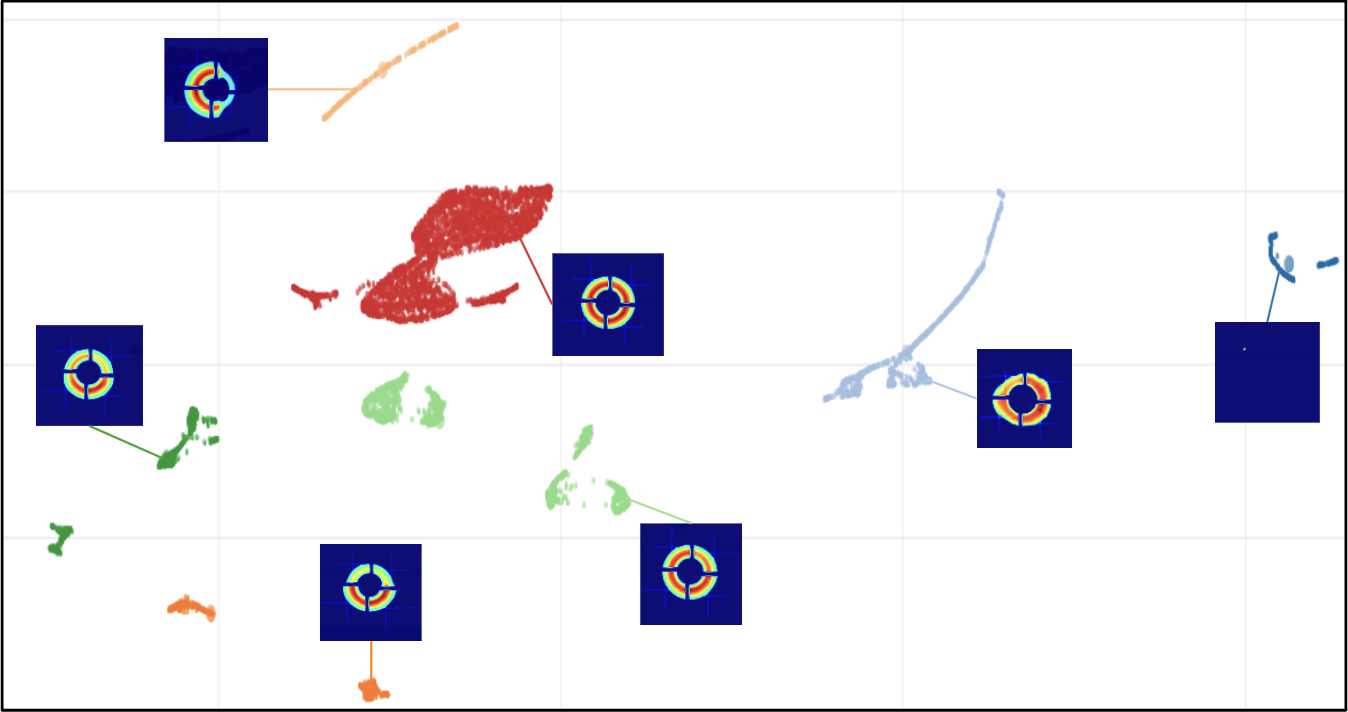
Matrix Sketching for Online Analysis of LCLS Imaging Datasets
SuperComputing (2024)
X-ray light source facilities such as the Linac Coherence Light Source (LCLS) at SLAC National Accelerator Laboratory generate massive amounts of data that need to be analyzed quickly to inform ongoing experiments. The analysis of data streams coming from various parts of the instrument has potential to feed back into instrument operation or experiment steering. For example, shot-to-shot images of the beam profile inform on the quality of the beam delivery while downstream data read from large area detectors inform on the state of diffraction experiments carried on samples of interests at various beamlines. However, the high repetition rate and high dimensionality of these data streams make their analysis challenging, both in terms of scalability and interpretability. In this work, we propose an image monitoring and classification framework that follows a three-stage process: dimensionality reduction using principal component analysis on a matrix sketch, visualization using UMAP, and clustering using OPTICS. In the dimensionality reduction step, we combine the Priority Sampling algorithm with a modified Frequent Directions algorithm to produce a rank-adaptive accelerated matrix sketching (ARAMS) algorithm, wherein practitioners specify the target error of the sketch as opposed to the rank. Furthermore, the framework is parallel, enabling real-time analysis of the underpinning structure of the data. This framework demonstrates strong empirical performance and scalability. We explore its effectiveness on both beam profile data and diffraction data from recent LCLS experiments.
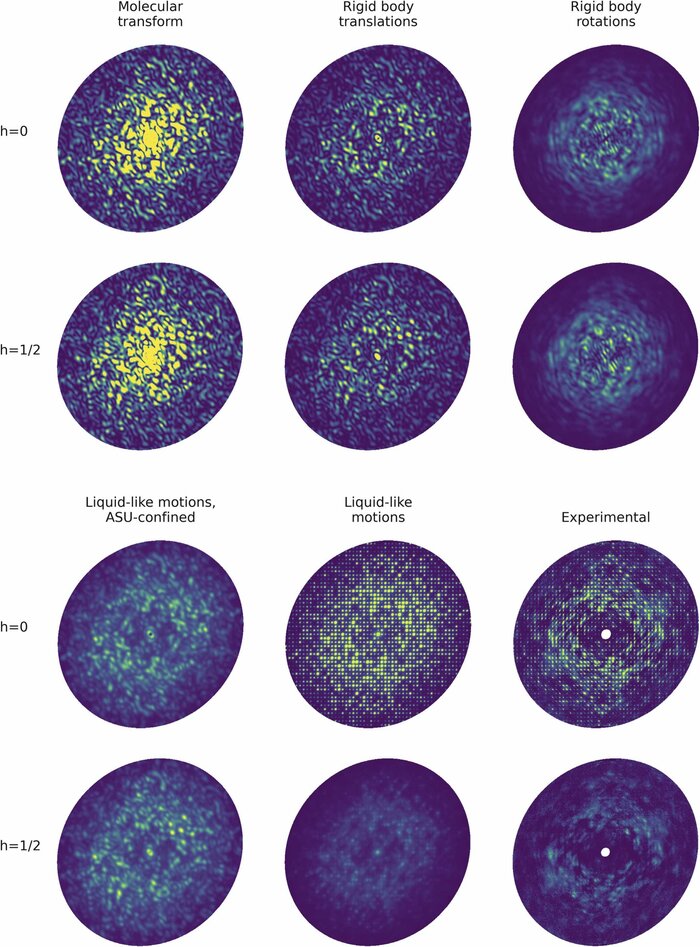
Modeling diffuse scattering with simple, physically interpretable models
Methods in enzymology (2023)
Diffuse scattering has long been proposed to probe protein dynamics relevant for biological function, and more recently, as a tool to aid structure determination. Despite recent advances in measuring and modeling this signal, the field has not been able to routinely use experimental diffuse scattering for either application. A persistent challenge has been to devise models that are sophisticated enough to robustly reproduce experimental diffuse features but remain readily interpretable from the standpoint of structural biology. This chapter presents eryx, a suite of computational tools to evaluate the primary models of disorder that have been used to analyze protein diffuse scattering. By facilitating comparative modeling, eryx aims to provide insights into the physical origins of this signal and help identify the sources of disorder that are critical for reproducing experimental features. This framework also lays the groundwork for the development of more advanced models that integrate different types of disorder without loss of interpretability.
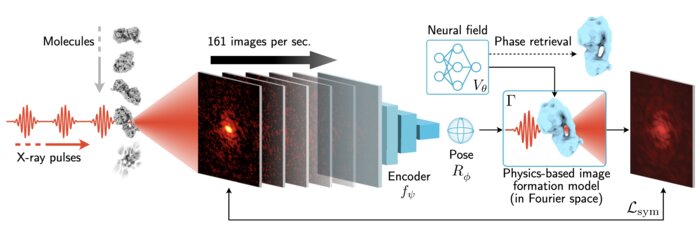
Scalable 3D Reconstruction From Single Particle X-Ray Diffraction Images Based on Online Machine Learning
arXiv preprint (2023)
X-ray free-electron lasers (XFELs) offer unique capabilities for measuring the structure and dynamics of biomolecules, helping us understand the basic building blocks of life. Notably, high-repetition-rate XFELs enable single particle imaging (X-ray SPI) where individual, weakly scattering biomolecules are imaged under near-physiological conditions with the opportunity to access fleeting states that cannot be captured in cryogenic or crystallized conditions. Existing X-ray SPI reconstruction algorithms, which estimate the unknown orientation of a particle in each captured image as well as its shared 3D structure, are inadequate in handling the massive datasets generated by these emerging XFELs. Here, we introduce X-RAI, an online reconstruction framework that estimates the structure of a 3D macromolecule from large X-ray SPI datasets. X-RAI consists of a convolutional encoder, which amortizes pose estimation over large datasets, as well as a physics-based decoder, which employs an implicit neural representation to enable high-quality 3D reconstruction in an end-to-end, self-supervised manner. We demonstrate that X-RAI achieves state-of-the-art performance for small-scale datasets in simulation and challenging experimental settings and demonstrate its unprecedented ability to process large datasets containing millions of diffraction images in an online fashion. These abilities signify a paradigm shift in X-ray SPI towards real-time capture and reconstruction.
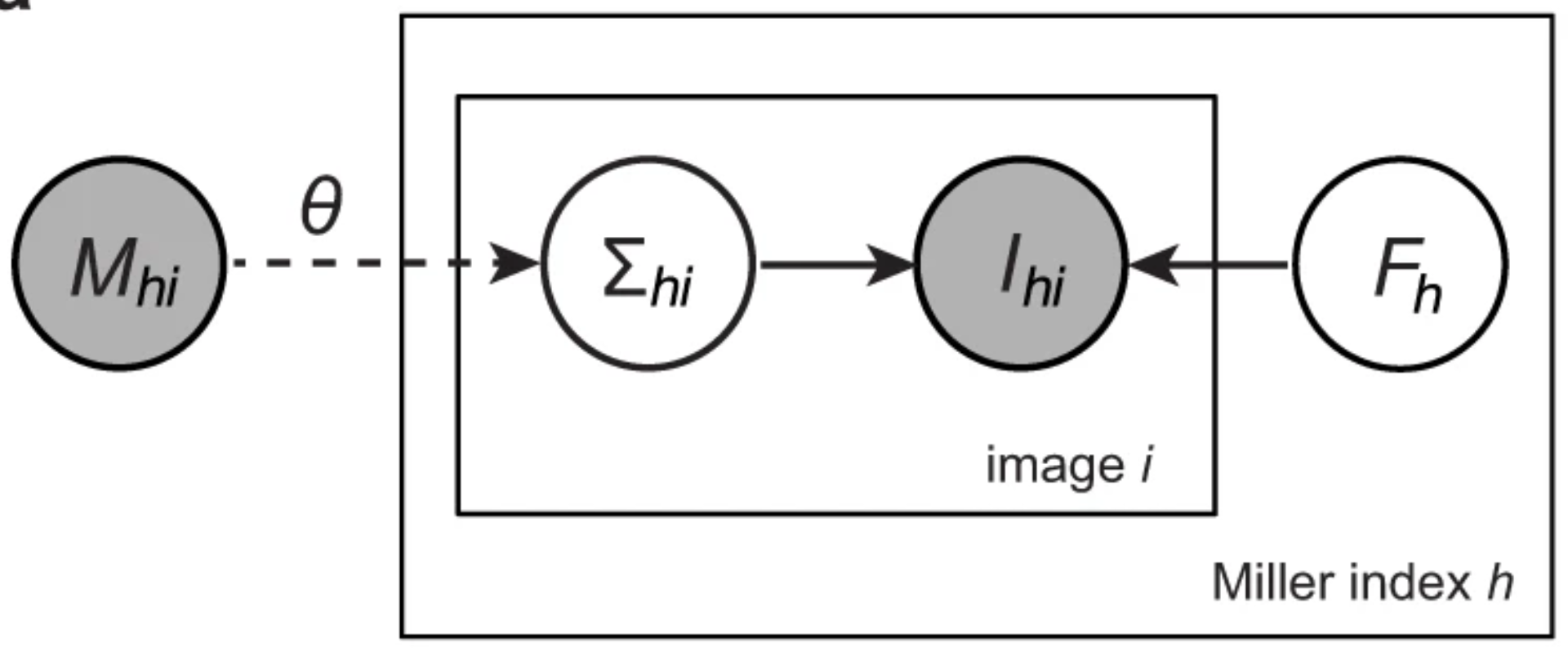
A unifying Bayesian framework for merging X-ray diffraction data
Nature Communications (2022)
Novel X-ray methods are transforming the study of the functional dynamics of biomolecules. Key to this revolution is detection of often subtle conformational changes from diffraction data. Diffraction data contain patterns of bright spots known as reflections. To compute the electron density of a molecule, the intensity of each reflection must be estimated, and redundant observations reduced to consensus intensities. Systematic effects, however, lead to the measurement of equivalent reflections on different scales, corrupting observation of changes in electron density. Here, we present a modern Bayesian solution to this problem, which uses deep learning and variational inference to simultaneously rescale and merge reflection observations. We successfully apply this method to monochromatic and polychromatic single-crystal diffraction data, as well as serial femtosecond crystallography data. We find that this approach is applicable to the analysis of many types of diffraction experiments, while accurately and sensitively detecting subtle dynamics and anomalous scattering.
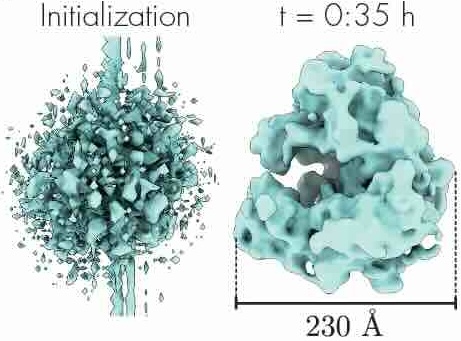
CryoAI: Amortized inference of poses for ab initio reconstruction of 3d molecular volumes from real cryo-em images
European Conference on Computer Vision (2022)
Cryo-electron microscopy (cryo-EM) has become a tool of fundamental importance in structural biology, helping us understand the basic building blocks of life. The algorithmic challenge of cryo-EM is to jointly estimate the unknown 3D poses and the 3D electron scattering potential of a biomolecule from millions of extremely noisy 2D images. Existing reconstruction algorithms, however, cannot easily keep pace with the rapidly growing size of cryo-EM datasets due to their high computational and memory cost. We introduce cryoAI, an ab initio reconstruction algorithm for homogeneous conformations that uses direct gradient-based optimization of particle poses and the electron scattering potential from single-particle cryo-EM data. CryoAI combines a learned encoder that predicts the poses of each particle image with a physics-based decoder to aggregate each particle image into an implicit representation of the scattering potential volume. This volume is stored in the Fourier domain for computational efficiency and leverages a modern coordinate network architecture for memory efficiency. Combined with a symmetrized loss function, this framework achieves results of a quality on par with state-of-the-art cryo-EM solvers for both simulated and experimental data, one order of magnitude faster for large datasets and with significantly lower memory requirements than existing methods.