DynaBoost
Selective Enhancement and Interpretation of X-ray Scattering Signals from Protein Crystals under Terahertz Excitation
Frédéric Poitevin , Oliver Hoidn , Alec Follmer
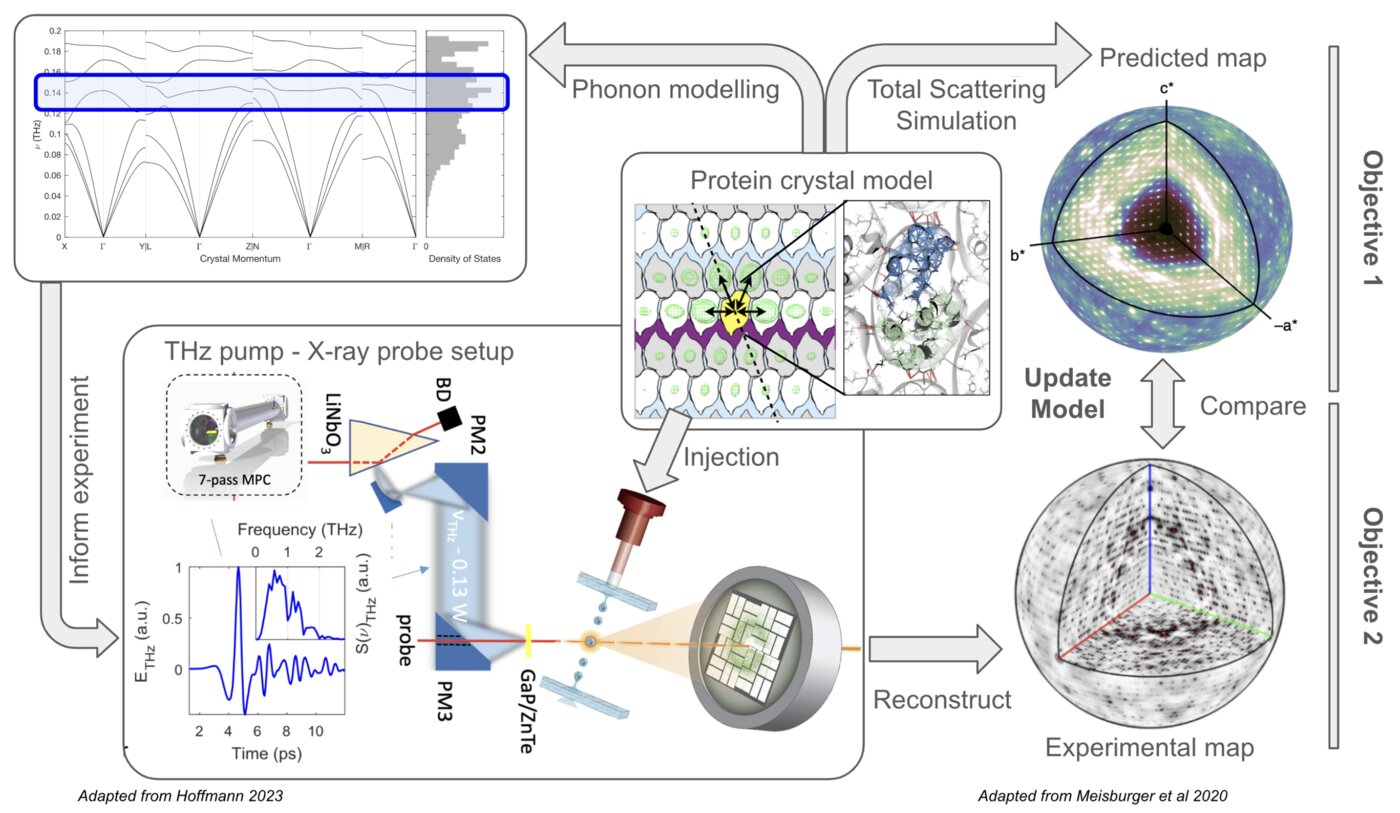
Project Goal
The goal of this project is to establish a scientific program focused on studying enzyme dynamics using terahertz (THz) capabilities emerging at SLAC. It will develop a combined approach to simulating, measuring, and analyzing the scattering signal arising from THz-driven perturbations of vibrational protein motions in the crystal lattice context. Computational models will inform the design and implementation of THz-pump X-ray diffraction probe techniques at LCLS-II-HE, complementing experimental efforts underway at SSRL. Data analysis pipelines will be developed to efficiently reconstruct 3D scattering maps that will readily inform on specific structural motions at the molecular level.
Through this proposed research, we anticipate uncovering new insights into the structural dynamics of proteins, shedding light on the fundamental processes underlying enzymatic catalysis and protein function. Specifically, exciting hypotheses in the area of protein dynamics are emerging where global collective motions of proteins and/or local electric fields within enzyme active site reaction centers may be the dominant factors that facilitate their catalytic abilities and are, in principle, at play in many protein systems. Both global collective motions and electric field effects can be studied using ultrafast excitations in the terahertz (THz) frequency range (⇠ 3 � 300 cm�1) providing new opportunities to expand the scope and study of enzymatic mechanisms and protein dynamics with time-resolved THz capabilities offered at SLAC. While computational and experimental support for these ideas is beginning to emerge, direct observation and quantification of these effects would represent a paradigm shift in the field of enzymology. In anticipation of these experimental capabilities at LCLS-II, this project focuses on the development and utilization of a computational framework for simulating time-resolved THz-pump X-ray diffraction experiments on protein crystals.
Recent advancements in the generation of high peak-field sources of few-cycle terahertz (THz) pulses have opened new avenues for exploring dynamic processes in solids, where the combination of single-cycle THz excitation with ultrafast X-ray diffraction probe pulses allows for the direct tracking of atomic displacements within the unit cell when driven by an intense electromagnetic field. While this type of approach has shown its potential upon application to ferroelectric crystals at LCLS, protein motions are inherently much more complex, involving non-trivial molecular re- arrangements in more porous and hydrated environments. This complexity presents a significant challenge to studying THz-induced motions that we address through a simulation-forward first-principles based approach to experimental design and data analysis.
Moreover, development of a theoretical basis for these experiments will significantly aid in the interpretation and clarification of on-going diffuse X-ray scattering analyses in single-crystal protein crystallography experiments, which is a challenging and active area of investigation for studying protein dynamics. By correlating experimental observations with simulations of protein structures and their dynamics, our approach will help refine the interpretation of diffuse X-ray scattering patterns, providing enhanced clarity and precision in elucidating the structural dynamics of proteins within crystalline environments.
References
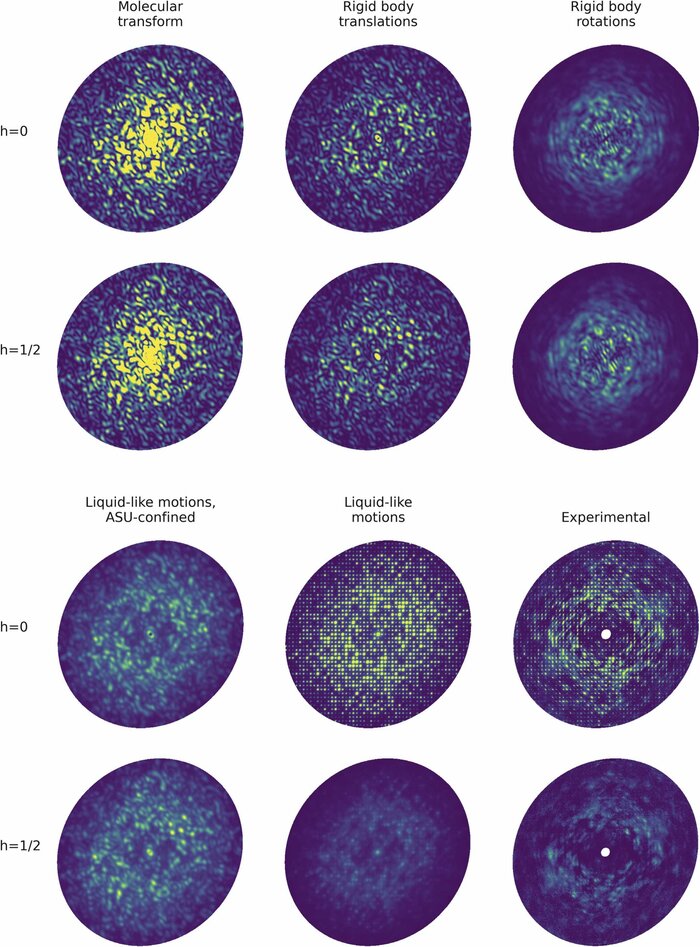
Modeling diffuse scattering with simple, physically interpretable models
Methods in enzymology (2023)
Diffuse scattering has long been proposed to probe protein dynamics relevant for biological function, and more recently, as a tool to aid structure determination. Despite recent advances in measuring and modeling this signal, the field has not been able to routinely use experimental diffuse scattering for either application. A persistent challenge has been to devise models that are sophisticated enough to robustly reproduce experimental diffuse features but remain readily interpretable from the standpoint of structural biology. This chapter presents eryx, a suite of computational tools to evaluate the primary models of disorder that have been used to analyze protein diffuse scattering. By facilitating comparative modeling, eryx aims to provide insights into the physical origins of this signal and help identify the sources of disorder that are critical for reproducing experimental features. This framework also lays the groundwork for the development of more advanced models that integrate different types of disorder without loss of interpretability.
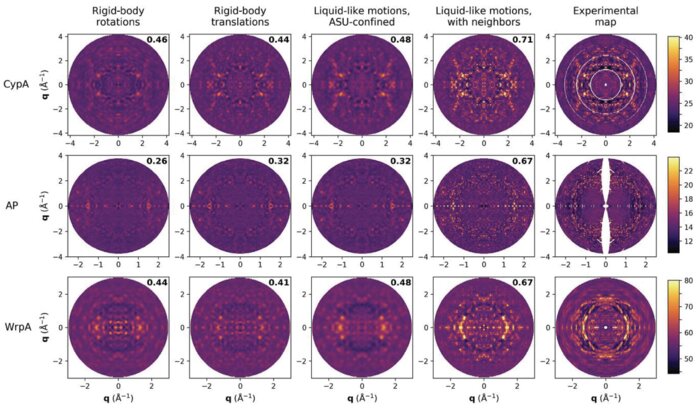
Intermolecular correlations are necessary to explain diffuse scattering from protein crystals
IUCrJ (2018)
Conformational changes drive protein function, including catalysis, allostery and signaling. X-ray diffuse scattering from protein crystals has frequently been cited as a probe of these correlated motions, with significant potential to advance our understanding of biological dynamics. However, recent work has challenged this prevailing view, suggesting instead that diffuse scattering primarily originates from rigid-body motions and could therefore be applied to improve structure determination. To investigate the nature of the disorder giving rise to diffuse scattering, and thus the potential applications of this signal, a diverse repertoire of disorder models was assessed for its ability to reproduce the diffuse signal reconstructed from three protein crystals. This comparison revealed that multiple models of intramolecular conformational dynamics, including ensemble models inferred from the Bragg data, could not explain the signal. Models of rigid-body or short-range liquid-like motions, in which dynamics are confined to the biological unit, showed modest agreement with the diffuse maps, but were unable to reproduce experimental features indicative of long-range correlations. Extending a model of liquid-like motions to include disorder across neighboring proteins in the crystal significantly improved agreement with all three systems and highlighted the contribution of intermolecular correlations to the observed signal. These findings anticipate a need to account for intermolecular disorder in order to advance the interpretation of diffuse scattering to either extract biological motions or aid structural inference.