Publications
2025
- cryoEM
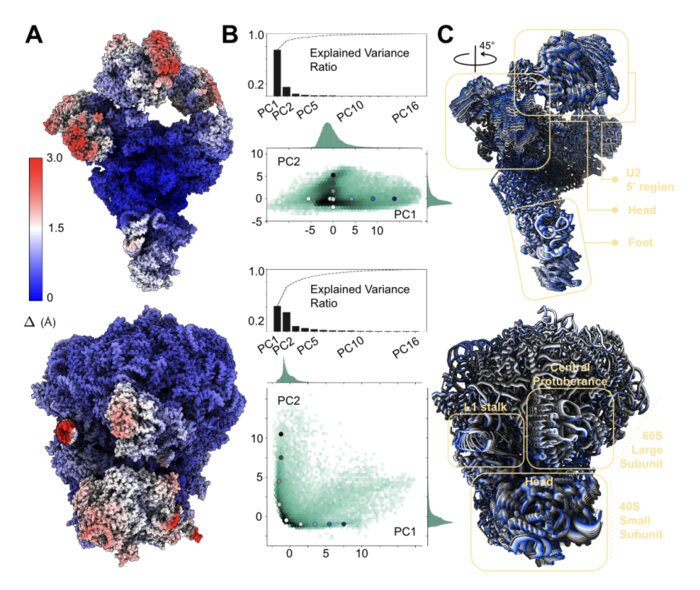 Unsupervised learning of structural variability in cryo-EM data using normal mode analysis of deformable atomic modelsYoussef Nashed, Julien Martel, Ariana Peck, and 6 more authorsbioRxiv, Jul 2025
Unsupervised learning of structural variability in cryo-EM data using normal mode analysis of deformable atomic modelsYoussef Nashed, Julien Martel, Ariana Peck, and 6 more authorsbioRxiv, Jul 2025Cryogenic electron microscopy (cryo-EM) has emerged as the method of choice to characterize the structural variability of biomolecules at near-atomic resolution. We present a reconstruction approach that eliminates the need for post-hoc atomic model fitting in 3D maps by deforming a given atomic model along its normal modes directly against the 2D data. This end-to-end approach inherently reduces the risk of error propagation while increasing interpretability of resulting structural ensembles.
- SPI
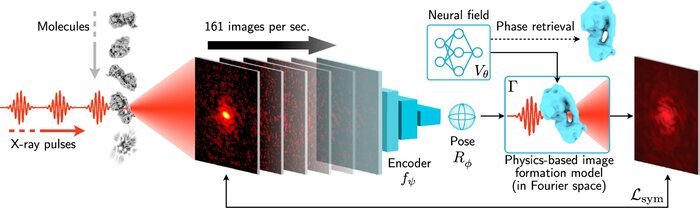 Scalable 3D reconstruction for X-ray single particle imaging with online machine learningJay Shenoy, Axel Levy, Kartik Ayyer, and 2 more authorsNature Communications, Jul 2025
Scalable 3D reconstruction for X-ray single particle imaging with online machine learningJay Shenoy, Axel Levy, Kartik Ayyer, and 2 more authorsNature Communications, Jul 2025X-ray free-electron lasers offer unique capabilities for measuring the structure and dynamics of biomolecules, helping us understand the basic building blocks of life. Notably, high-repetition-rate free-electron lasers enable single particle imaging, where individual, weakly scattering biomolecules are imaged under near-physiological conditions with the opportunity to access fleeting states that cannot be captured in cryogenic or crystallized conditions. Existing X-ray single particle reconstruction algorithms, which estimate the particle orientation for each image independently, are slow and memory-intensive when handling the massive datasets generated by emerging free-electron lasers. Here, we introduce X-RAI (X-Ray single particle imaging with Amortized Inference), an online reconstruction framework that estimates the structure of 3D macromolecules from large X-ray single particle datasets. X-RAI consists of a convolutional encoder, which amortizes pose estimation over large datasets, as well as a physics-based decoder, which employs an implicit neural representation to enable high-quality 3D reconstruction in an end-to-end, self-supervised manner. We demonstrate that X-RAI achieves state-of-the-art performance for small-scale datasets in simulation and challenging experimental settings and demonstrate its unprecedented ability to process large datasets containing millions of diffraction images in an online fashion. These abilities signify a paradigm shift in X-ray single particle imaging towards real-time reconstruction.
- CryoEM
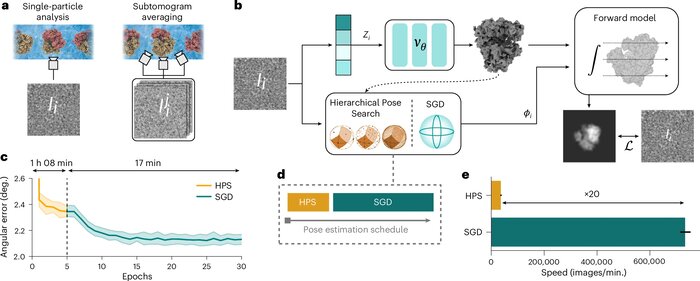 CryoDRGN-AI: neural ab initio reconstruction of challenging cryo-EM and cryo-ET datasetsAxel Levy, Rishwanth Raghu, J Ryan Feathers, and 7 more authorsNature Methods, Jun 2025
CryoDRGN-AI: neural ab initio reconstruction of challenging cryo-EM and cryo-ET datasetsAxel Levy, Rishwanth Raghu, J Ryan Feathers, and 7 more authorsNature Methods, Jun 2025Proteins and other biomolecules form dynamic macromolecular machines that are tightly orchestrated to move, bind and perform chemistry. Cryo-electron microscopy and cryo-electron tomography can access the intrinsic heterogeneity of these complexes and are therefore key tools for understanding their function. However, three-dimensional reconstruction of the collected imaging data presents a challenging computational problem, especially without any starting information, a setting termed ab initio reconstruction. Here we introduce cryoDRGN-AI, a method leveraging an expressive neural representation and combining an exhaustive search strategy with gradient-based optimization to process challenging heterogeneous datasets. Using cryoDRGN-AI, we reveal new conformational states in large datasets, reconstruct previously unresolved motions from unfiltered datasets and demonstrate ab initio reconstruction of biomolecular complexes from in situ data. With this expressive and scalable model for structure determination, we hope to unlock the full potential of cryo-electron microscopy and cryo-electron tomography as a high-throughput tool for structural biology and discovery.
- Crystallography
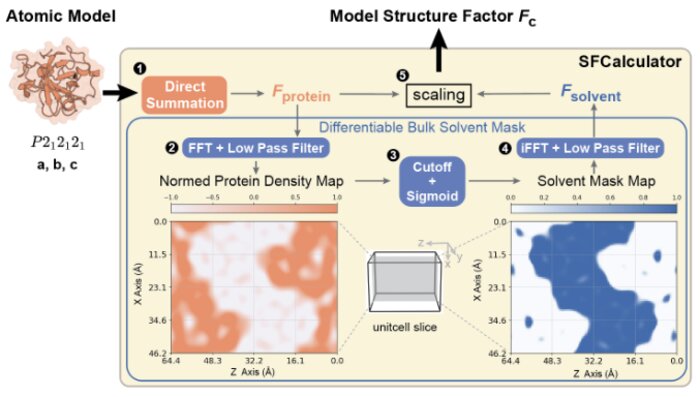 SFCalculator: connecting deep generative models and crystallographyMinhuan Li, Kevin Dalton, and Doeke HekstrabioRxiv, Jan 2025
SFCalculator: connecting deep generative models and crystallographyMinhuan Li, Kevin Dalton, and Doeke HekstrabioRxiv, Jan 2025Proteins drive biochemical transformations by transitioning through distinct conformational states. Understanding these states is essential for modulating protein function. Although X-ray crystallography has enabled revolutionary advances in protein structure prediction by machine learning, this connection was made at the level of atomic models, not the underlying data. This lack of connection to crystallographic data limits the potential for further advances in both the accuracy of protein structure prediction and the application of machine learning to experimental structure determination. Here, we present SFCalculator, a differentiable pipeline that generates crystallographic observables from atomistic molecular structures with bulk solvent correction, bridging crystallographic data and neural network-based molecular modeling. We validate SFCalculator against conventional methods and demonstrate its utility by establishing three important proof-of-concept applications. First, SFCalculator enables accurate placement of molecular models relative to crystal lattices (known as phasing). Second, SFCalculator enables the search of the latent space of generative models for conformations that fit crystallographic data and are, therefore, also implicitly constrained by the information encoded by the model. Finally, SFCalculator enables the use of crystallographic data during training of generative models, enabling these models to generate an ensemble of conformations consistent with crystallographic data. SFCalculator, therefore, enables a new generation of analytical paradigms integrating crystallographic data and machine learning.




2024
- Data Reduction
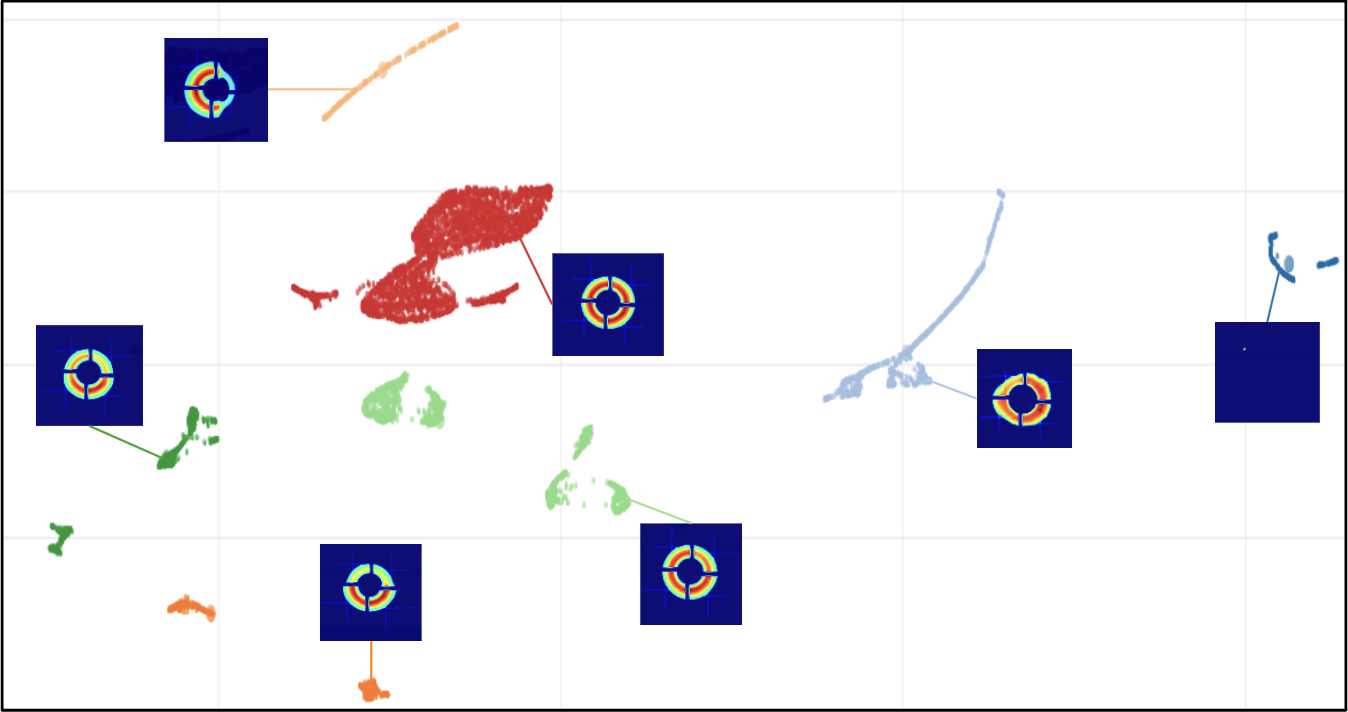 Matrix Sketching for Online Analysis of LCLS Imaging DatasetsJohn Winnicki, Frédéric Poitevin, Haoyuan Li, and 1 more authorIn SuperComputing, Jan 2024
Matrix Sketching for Online Analysis of LCLS Imaging DatasetsJohn Winnicki, Frédéric Poitevin, Haoyuan Li, and 1 more authorIn SuperComputing, Jan 2024X-ray light source facilities such as the Linac Coherence Light Source (LCLS) at SLAC National Accelerator Laboratory generate massive amounts of data that need to be analyzed quickly to inform ongoing experiments. The analysis of data streams coming from various parts of the instrument has potential to feed back into instrument operation or experiment steering. For example, shot-to-shot images of the beam profile inform on the quality of the beam delivery while downstream data read from large area detectors inform on the state of diffraction experiments carried on samples of interests at various beamlines. However, the high repetition rate and high dimensionality of these data streams make their analysis challenging, both in terms of scalability and interpretability. In this work, we propose an image monitoring and classification framework that follows a three-stage process: dimensionality reduction using principal component analysis on a matrix sketch, visualization using UMAP, and clustering using OPTICS. In the dimensionality reduction step, we combine the Priority Sampling algorithm with a modified Frequent Directions algorithm to produce a rank-adaptive accelerated matrix sketching (ARAMS) algorithm, wherein practitioners specify the target error of the sketch as opposed to the rank. Furthermore, the framework is parallel, enabling real-time analysis of the underpinning structure of the data. This framework demonstrates strong empirical performance and scalability. We explore its effectiveness on both beam profile data and diffraction data from recent LCLS experiments.
- Crystallography
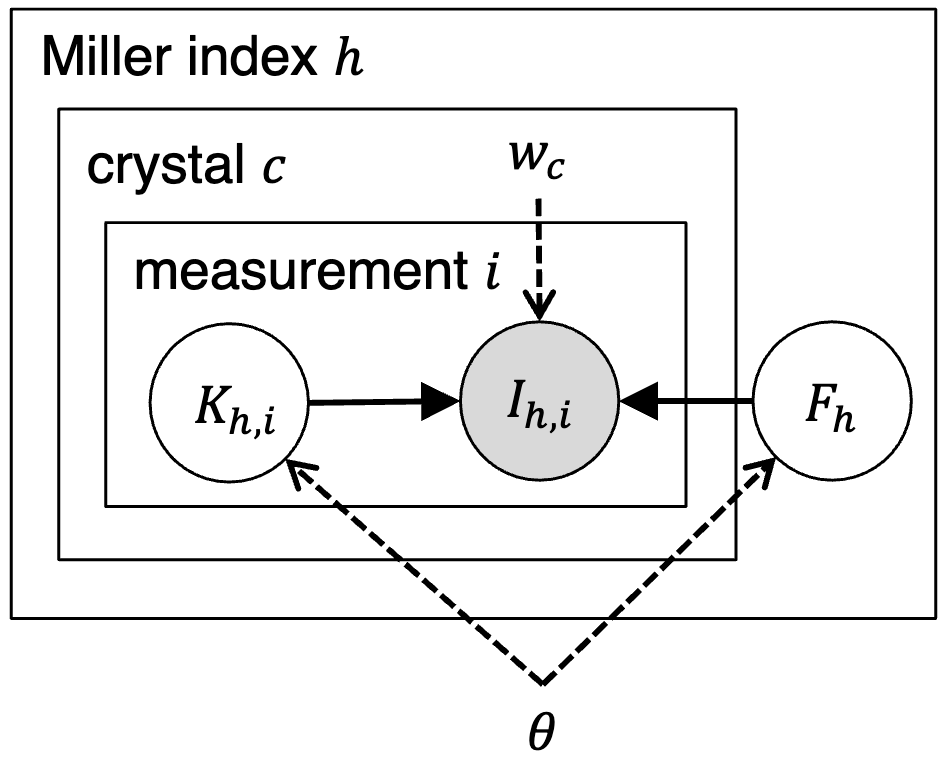 Assessing the applicability of Bayesian inference for merging small molecule microED dataHuanghao Mai, Ariana Peck, Kevin M Dalton, and 4 more authorsChemRxiv, Jan 2024
Assessing the applicability of Bayesian inference for merging small molecule microED dataHuanghao Mai, Ariana Peck, Kevin M Dalton, and 4 more authorsChemRxiv, Jan 2024Microcrystal electron diffraction (MicroED) is an emerging technique for characterizing small molecule structures from nanoscale crystals. Merging data from multiple crystals is a particularly challenging step in the microED workflow. A common practice is to manually curate datasets and apply scaling programs conventionally utilized in rotational X-ray diffraction (XRD), but this could be time-consuming and risks introducing human bias in data analysis. Recently, a Bayesian inference program named Careless (Dalton et al., 2022) has demonstrated excellent performance in merging macromolecular XRD data. Here, the applicability of Careless to small molecule microED data is evaluated and an investigation of the impact of dataset curation is performed. Benchmarking against XDS/XSCALE shows that Careless is an effective complementary approach that merges data to a higher CC1/2 value at high resolution. Furthermore, merging outcomes are not significantly improved by curating datasets either manually or with an automated extension to Careless, cautioning against the common practice of manual dataset curation.
- Interpretability
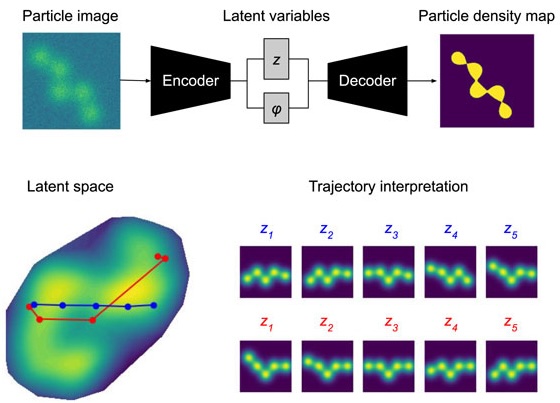 Towards interpretable Cryo-EM: disentangling latent spaces of molecular conformationsDavid A Klindt, Aapo Hyvärinen, Axel Levy, and 2 more authorsFrontiers in Molecular Biosciences, Jan 2024
Towards interpretable Cryo-EM: disentangling latent spaces of molecular conformationsDavid A Klindt, Aapo Hyvärinen, Axel Levy, and 2 more authorsFrontiers in Molecular Biosciences, Jan 2024Molecules are essential building blocks of life and their different conformations (i.e., shapes) crucially determine the functional role that they play in living organisms. Cryogenic Electron Microscopy (cryo-EM) allows for acquisition of large image datasets of individual molecules. Recent advances in computational cryo-EM have made it possible to learn latent variable models of conformation landscapes. However, interpreting these latent spaces remains a challenge as their individual dimensions are often arbitrary. The key message of our work is that this interpretation challenge can be viewed as an Independent Component Analysis (ICA) problem where we seek models that have the property of identifiability. That means, they have an essentially unique solution, representing a conformational latent space that separates the different degrees of freedom a molecule is equipped with in nature. Thus, we aim to advance the computational field of cryo-EM beyond visualizations as we connect it with the theoretical framework of (nonlinear) ICA and discuss the need for identifiable models, improved metrics, and benchmarks. Moving forward, we propose future directions for enhancing the disentanglement of latent spaces in cryo-EM, refining evaluation metrics and exploring techniques that leverage physics-based decoders of biomolecular systems. Moreover, we discuss how future technological developments in time-resolved single particle imaging may enable the application of nonlinear ICA models that can discover the true conformation changes of molecules in nature. The pursuit of interpretable conformational latent spaces will empower researchers to unravel complex biological processes and facilitate targeted interventions. This has significant implications for drug discovery and structural biology more broadly. More generally, latent variable models are deployed widely across many scientific disciplines. Thus, the argument we present in this work has much broader applications in AI for science if we want to move from impressive nonlinear neural network models to mathematically grounded methods that can help us learn something new about nature.
- Crystallography
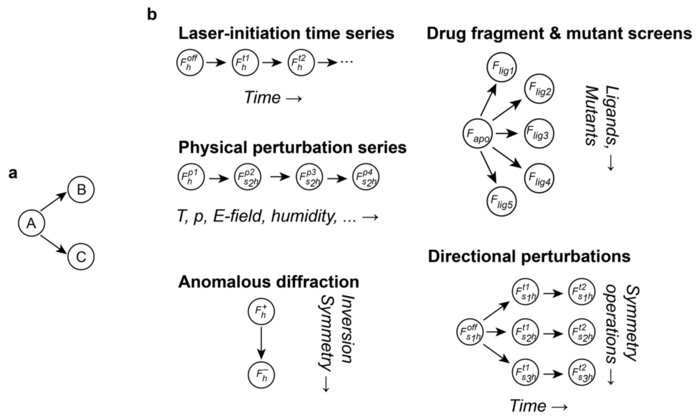 Sensitive Detection of Structural Differences using a Statistical Framework for Comparative CrystallographyDoeke R. Hekstra, Harrison K. Wang, Margaret A. Klureza, and 2 more authorsbioRxiv, Jul 2024
Sensitive Detection of Structural Differences using a Statistical Framework for Comparative CrystallographyDoeke R. Hekstra, Harrison K. Wang, Margaret A. Klureza, and 2 more authorsbioRxiv, Jul 2024Chemical and conformational changes underlie the functional cycles of proteins. Comparative crystallography can reveal these changes over time, over ligands, and over chemical and physical perturbations in atomic detail. A key difficulty, however, is that the resulting observations must be placed on the same scale by correcting for experimental factors. We recently introduced a Bayesian framework for correcting (scaling) X-ray diffraction data by combining deep learning with statistical priors informed by crystallographic theory. To scale comparative crystallography data, we here combine this framework with a multivariate statistical theory of comparative crystallography. By doing so, we find strong improvements in the detection of protein dynamics, element-specific anomalous signal, and the binding of drug fragments.
- Crystallography
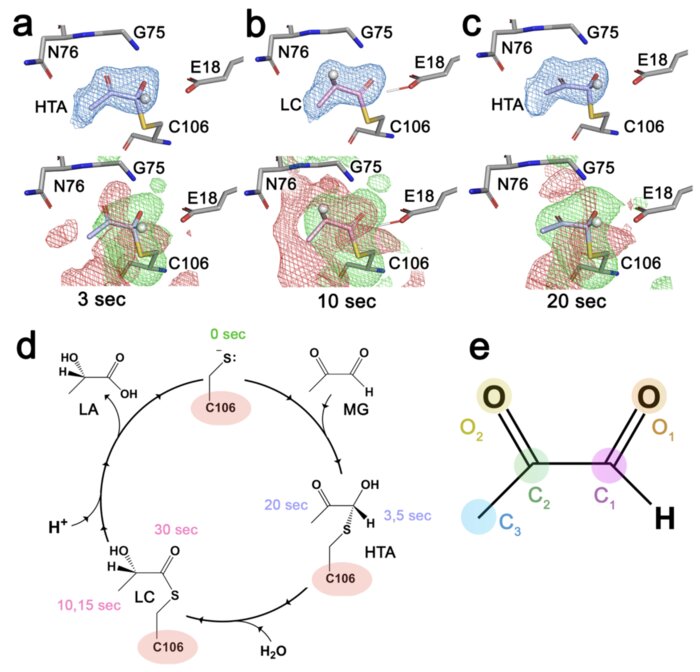 Resolving DJ-1 Glyoxalase Catalysis Using Mix-and-Inject Serial Crystallography at a SynchrotronKara A. Zielinski, Cole Dolamore, Kevin M. Dalton, and 7 more authorsbioRxiv, Jul 2024
Resolving DJ-1 Glyoxalase Catalysis Using Mix-and-Inject Serial Crystallography at a SynchrotronKara A. Zielinski, Cole Dolamore, Kevin M. Dalton, and 7 more authorsbioRxiv, Jul 2024DJ-1 (PARK7) is an intensively studied protein whose cytoprotective activities are dysregulated in multiple diseases. DJ-1 has been reported as having two distinct enzymatic activities in defense against reactive carbonyl species that are difficult to distinguish in conventional biochemical experiments. Here, we establish the mechanism of DJ-1 using a synchrotron-compatible version of mix-and-inject-serial crystallography (MISC), which was previously performed only at XFELs, to directly observe DJ-1 catalysis. We designed and used new diffusive mixers to collect time-resolved Laue diffraction data of DJ-1 catalysis at a pink beam synchrotron beamline. Analysis of structurally similar methylglyoxal-derived intermediates formed through the DJ-1 catalytic cycle shows that the enzyme catalyzes nearly two turnovers in the crystal and defines key aspects of its glyoxalase mechanism. In addition, DJ-1 shows allosteric communication between a distal site at the dimer interface and the active site that changes during catalysis. Our results rule out the widely cited deglycase mechanism for DJ-1 action and provide an explanation for how DJ-1 produces L-lactate with high chiral purity.
- Crystallography
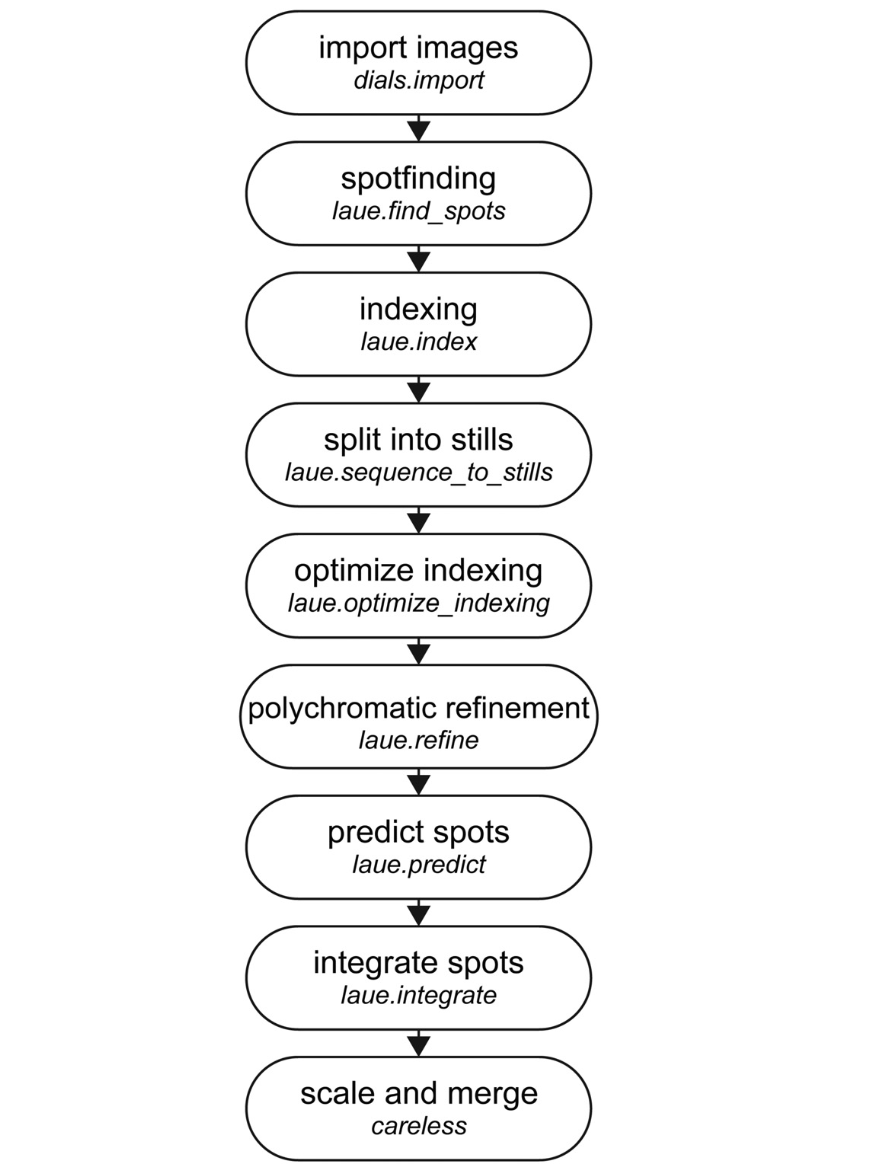 Laue-DIALS: Open-source software for polychromatic x-ray diffraction dataRick A. Hewitt*, Kevin M. Dalton*, Derek A. Mendez, and 9 more authorsStructural Dynamics, Oct 2024
Laue-DIALS: Open-source software for polychromatic x-ray diffraction dataRick A. Hewitt*, Kevin M. Dalton*, Derek A. Mendez, and 9 more authorsStructural Dynamics, Oct 2024Most x-ray sources are inherently polychromatic. Polychromatic (“pink”) x-rays provide an efficient way to conduct diffraction experiments as many more photons can be used and large regions of reciprocal space can be probed without sample rotation during exposure—ideal conditions for time-resolved applications. Analysis of such data is complicated, however, causing most x-ray facilities to discard >99% of x-ray photons to obtain monochromatic data. Key challenges in analyzing polychromatic diffraction data include lattice searching, indexing and wavelength assignment, correction of measured intensities for wavelength-dependent effects, and deconvolution of harmonics. We recently described an algorithm, Careless, that can perform harmonic deconvolution and correct measured intensities for variation in wavelength when presented with integrated diffraction intensities and assigned wavelengths. Here, we present Laue-DIALS, an open-source software pipeline that indexes and integrates polychromatic diffraction data. Laue-DIALS is based on the dxtbx toolbox, which supports the DIALS software commonly used to process monochromatic data. As such, Laue-DIALS provides many of the same advantages: an open-source, modular, and extensible architecture, providing a robust basis for future development. We present benchmark results showing that Laue-DIALS, together with Careless, provides a suitable approach to the analysis of polychromatic diffraction data, including for time-resolved applications.
- Crystallography
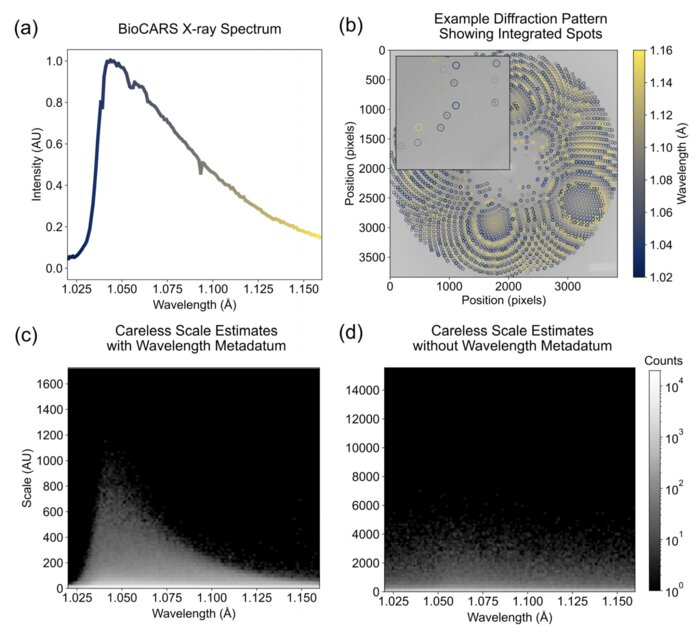 Scaling and merging time-resolved pink-beam diffraction with variational inferenceKara A. Zielinski, Cole Dolamore, Harrison K. Wang, and 6 more authorsStructural Dynamics, Nov 2024
Scaling and merging time-resolved pink-beam diffraction with variational inferenceKara A. Zielinski, Cole Dolamore, Harrison K. Wang, and 6 more authorsStructural Dynamics, Nov 2024Time-resolved x-ray crystallography (TR-X) at synchrotrons and free electron lasers is a promising technique for recording dynamics of molecules at atomic resolution. While experimental methods for TR-X have proliferated and matured, data analysis is often difficult. Extracting small, time-dependent changes in signal is frequently a bottleneck for practitioners. Recent work demonstrated this challenge can be addressed when merging redundant observations by a statistical technique known as variational inference (VI). However, the variational approach to time-resolved data analysis requires identification of successful hyperparameters in order to optimally extract signal. In this case study, we present a successful application of VI to time-resolved changes in an enzyme, DJ-1, upon mixing with a substrate molecule, methylglyoxal. We present a strategy to extract high signal-to-noise changes in electron density from these data. Furthermore, we conduct an ablation study, in which we systematically remove one hyperparameter at a time to demonstrate the impact of each hyperparameter choice on the success of our model. We expect this case study will serve as a practical example for how others may deploy VI in order to analyze their time-resolved diffraction data.
- Protein Folding
 Solving Inverse Problems in Protein Space Using Diffusion-Based PriorsAxel Levy, Eric R Chan, Sara Fridovich-Keil, and 3 more authorsarXiv preprint arXiv:2406.04239, Nov 2024
Solving Inverse Problems in Protein Space Using Diffusion-Based PriorsAxel Levy, Eric R Chan, Sara Fridovich-Keil, and 3 more authorsarXiv preprint arXiv:2406.04239, Nov 2024The interaction of a protein with its environment can be understood and controlled via its 3D structure. Experimental methods for protein structure determination, such as X-ray crystallography or cryogenic electron microscopy, shed light on biological processes but introduce challenging inverse problems. Learning-based approaches have emerged as accurate and efficient methods to solve these inverse problems for 3D structure determination, but are specialized for a predefined type of measurement. Here, we introduce a versatile framework to turn raw biophysical measurements of varying types into 3D atomic models. Our method combines a physics-based forward model of the measurement process with a pretrained generative model providing a task-agnostic, data-driven prior. Our method outperforms posterior sampling baselines on both linear and non-linear inverse problems. In particular, it is the first diffusion-based method for refining atomic models from cryo-EM density maps.
- CryoEM
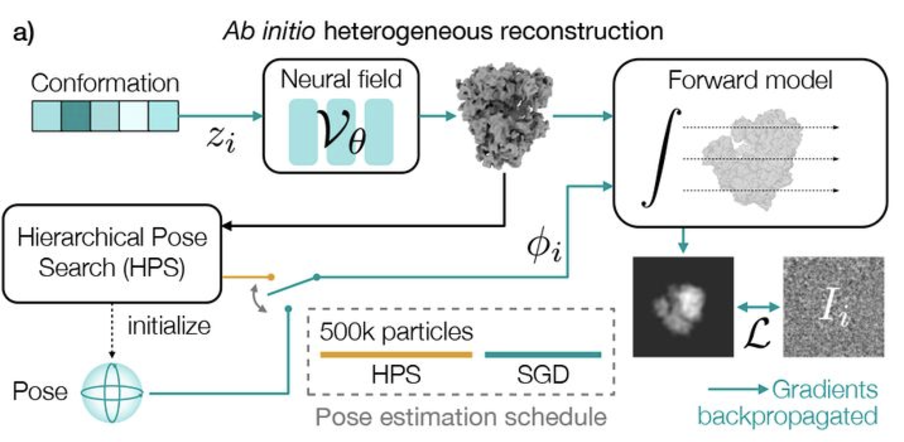 Revealing biomolecular structure and motion with neural ab initio cryo-EM reconstructionAxel Levy, Michal Grzadkowski, Frederic Poitevin, and 4 more authorsbioRxiv, Nov 2024
Revealing biomolecular structure and motion with neural ab initio cryo-EM reconstructionAxel Levy, Michal Grzadkowski, Frederic Poitevin, and 4 more authorsbioRxiv, Nov 2024Proteins and other biomolecules form dynamic macromolecular machines that are tightly orchestrated to move, bind, and perform chemistry. Cryo-electron microscopy (cryo-EM) can access the intrinsic heterogeneity of these complexes and is therefore a key tool for understanding mechanism and function. However, 3D reconstruction of the resulting imaging data presents a challenging computational problem, especially without any starting information, a setting termed ab initio reconstruction. Here, we introduce a method, DRGN-AI, for ab initio heterogeneous cryo-EM reconstruction. With a two-step hybrid approach combining search and gradient-based optimization, DRGN-AI can reconstruct dynamic protein complexes from scratch without input poses or initial models. Using DRGN-AI, we reconstruct the compositional and conformational variability contained in a variety of benchmark datasets, process an unfiltered dataset of the DSL1/SNARE complex fully ab initio, and reveal a new “supercomplex” state of the human erythrocyte ankyrin-1 complex. With this expressive and scalable model for structure determination, we hope to unlock the full potential of cryo-EM as a high-throughput tool for structural biology and discovery.
- Crystallography
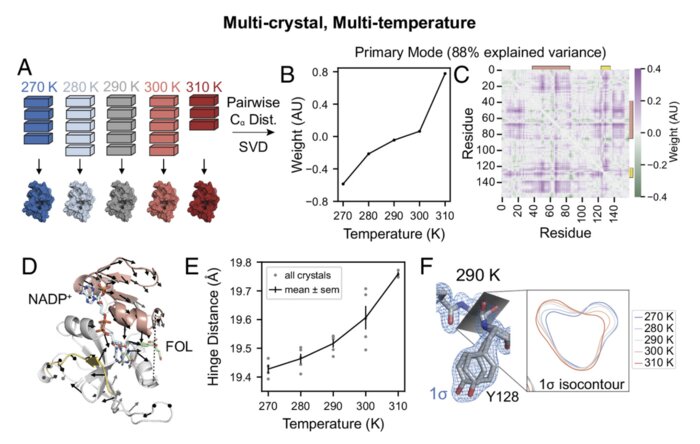 Perturbative diffraction methods resolve a conformational switch that facilitates a two-step enzymatic mechanismJack B. Greisman, Kevin M. Dalton, Dennis E. Brookner, and 6 more authorsNov 2024Publisher: Proceedings of the National Academy of Sciences
Perturbative diffraction methods resolve a conformational switch that facilitates a two-step enzymatic mechanismJack B. Greisman, Kevin M. Dalton, Dennis E. Brookner, and 6 more authorsNov 2024Publisher: Proceedings of the National Academy of SciencesEnzymes catalyze biochemical reactions through precise positioning of substrates, cofactors, and amino acids to modulate the transition-state free energy. However, the role of conformational dynamics remains poorly understood due to poor experimental access. This shortcoming is evident with Escherichia coli dihydrofolate reductase (DHFR), a model system for the role of protein dynamics in catalysis, for which it is unknown how the enzyme regulates the different active site environments required to facilitate proton and hydride transfer. Here, we describe ligand-, temperature-, and electric-field-based perturbations during X-ray diffraction experiments to map the conformational dynamics of the Michaelis complex of DHFR. We resolve coupled global and local motions and find that these motions are engaged by the protonated substrate to promote efficient catalysis. This result suggests a fundamental design principle for multistep enzymes in which pre-existing dynamics enable intermediates to drive rapid electrostatic reorganization to facilitate subsequent chemical steps.










2023
- Crystallography
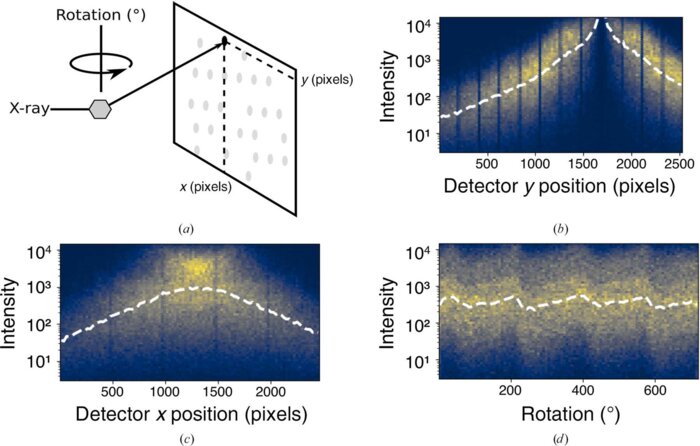 Correcting systematic errors in diffraction data with modern scaling algorithmsL. A. Aldama*, K. M. Dalton*, and D. R. HekstraNov 2023
Correcting systematic errors in diffraction data with modern scaling algorithmsL. A. Aldama*, K. M. Dalton*, and D. R. HekstraNov 2023X-ray diffraction enables the routine determination of the atomic structure of materials. Key to its success are data-processing algorithms that allow experimenters to determine the electron density of a sample from its diffraction pattern. Scaling, the estimation and correction of systematic errors in diffraction intensities, is an essential step in this process. These errors arise from sample heterogeneity, radiation damage, instrument limitations and other aspects of the experiment. New X-ray sources and sample-delivery methods, along with new experiments focused on changes in structure as a function of perturbations, have led to new demands on scaling algorithms. Classically, scaling algorithms use least-squares optimization to fit a model of common error sources to the observed diffraction intensities to force these intensities onto the same empirical scale. Recently, an alternative approach has been demonstrated which uses a Bayesian optimization method, variational inference, to simultaneously infer merged data along with corrections, or scale factors, for the systematic errors. Owing to its flexibility, this approach proves to be advantageous in certain scenarios. This perspective briefly reviews the history of scaling algorithms and contrasts them with variational inference. Finally, appropriate use cases are identified for the first such algorithm, Careless, guidance is offered on its use and some speculations are made about future variational scaling methods.
- SPI
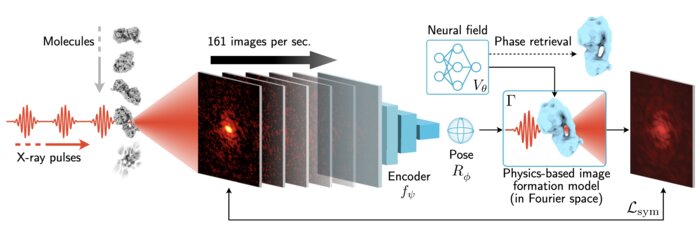 Scalable 3D Reconstruction From Single Particle X-Ray Diffraction Images Based on Online Machine LearningJay Shenoy, Axel Levy, Frédéric Poitevin, and 1 more authorarXiv preprint, Nov 2023
Scalable 3D Reconstruction From Single Particle X-Ray Diffraction Images Based on Online Machine LearningJay Shenoy, Axel Levy, Frédéric Poitevin, and 1 more authorarXiv preprint, Nov 2023X-ray free-electron lasers (XFELs) offer unique capabilities for measuring the structure and dynamics of biomolecules, helping us understand the basic building blocks of life. Notably, high-repetition-rate XFELs enable single particle imaging (X-ray SPI) where individual, weakly scattering biomolecules are imaged under near-physiological conditions with the opportunity to access fleeting states that cannot be captured in cryogenic or crystallized conditions. Existing X-ray SPI reconstruction algorithms, which estimate the unknown orientation of a particle in each captured image as well as its shared 3D structure, are inadequate in handling the massive datasets generated by these emerging XFELs. Here, we introduce X-RAI, an online reconstruction framework that estimates the structure of a 3D macromolecule from large X-ray SPI datasets. X-RAI consists of a convolutional encoder, which amortizes pose estimation over large datasets, as well as a physics-based decoder, which employs an implicit neural representation to enable high-quality 3D reconstruction in an end-to-end, self-supervised manner. We demonstrate that X-RAI achieves state-of-the-art performance for small-scale datasets in simulation and challenging experimental settings and demonstrate its unprecedented ability to process large datasets containing millions of diffraction images in an online fashion. These abilities signify a paradigm shift in X-ray SPI towards real-time capture and reconstruction.
- SPI
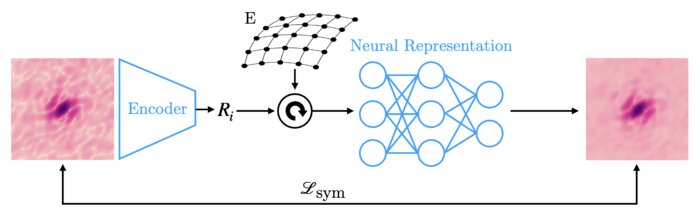 Amortized pose estimation for x-ray single particle imagingJay Shenoy, Axel Levy, Frédéric Poitevin, and 1 more authorIn Machine learning for structural biology Workshop, Nov 2023
Amortized pose estimation for x-ray single particle imagingJay Shenoy, Axel Levy, Frédéric Poitevin, and 1 more authorIn Machine learning for structural biology Workshop, Nov 2023X-ray single particle imaging (SPI) is a nascent technique that can capture the dynamics of biomolecules at room temperature. SPI experiments will one day collect tens of millions of images of the same molecule in order to overcome the weak scattering of individual proteins. Existing reconstruction algorithms will be unable to scale to datasets of this size because they perform computationally expensive search steps to estimate the orientation of the molecule in each image. In this work, we propose a reconstruction algorithm that amortizes the estimation of pose via an autoencoder framework. Our approach consists of a convolutional encoder that maps X-ray images to predicted poses and a physics-based decoder that implicitly fuses all the 2D scattering images into a volumetric representation of the molecule. We validate our method on 6 synthetic datasets of 2 distinct proteins, showing that for the largest datasets containing 5 million images, our technique can reconstruct the electron density in a single pass.
- CryoEM
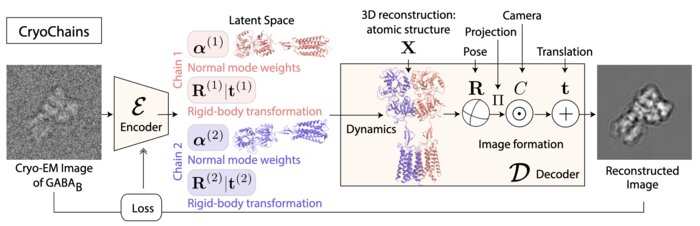 CryoChains: Heterogeneous Reconstruction of Molecular Assembly of Semi-flexible Chains from Cryo-EM ImagesBongjin Koo, Julien Martel, Ariana Peck, and 3 more authorsarXiv e-prints, Nov 2023
CryoChains: Heterogeneous Reconstruction of Molecular Assembly of Semi-flexible Chains from Cryo-EM ImagesBongjin Koo, Julien Martel, Ariana Peck, and 3 more authorsarXiv e-prints, Nov 2023Cryogenic electron microscopy (cryo-EM) has transformed structural biology by allowing to reconstruct 3D biomolecular structures up to nearatomic resolution. However, the 3D reconstruction process remains challenging, as the 3D structures may exhibit substantial shape variations, while the 2D image acquisition suffers from a low signal-to-noise ratio, requiring to acquire very large datasets that are time-consuming to process. Current reconstruction methods are precise but computationally expensive, or faster but lack a physically-plausible model of large molecular shape variations. To fill this gap, we propose CryoChains that encodes large deformations of biomolecules via rigid body transformation of their chains, while representing their finer shape variations with the normal mode analysis framework of biophysics. Our synthetic data experiments on the human GABAB and heat shock protein show that CryoChains gives a biophysicallygrounded quantification of the heterogeneous conformations of biomolecules, while reconstructing their 3D molecular structures at an improved resolution compared to the current fastest, interpretable deep learning method.
- Diffuse Scattering
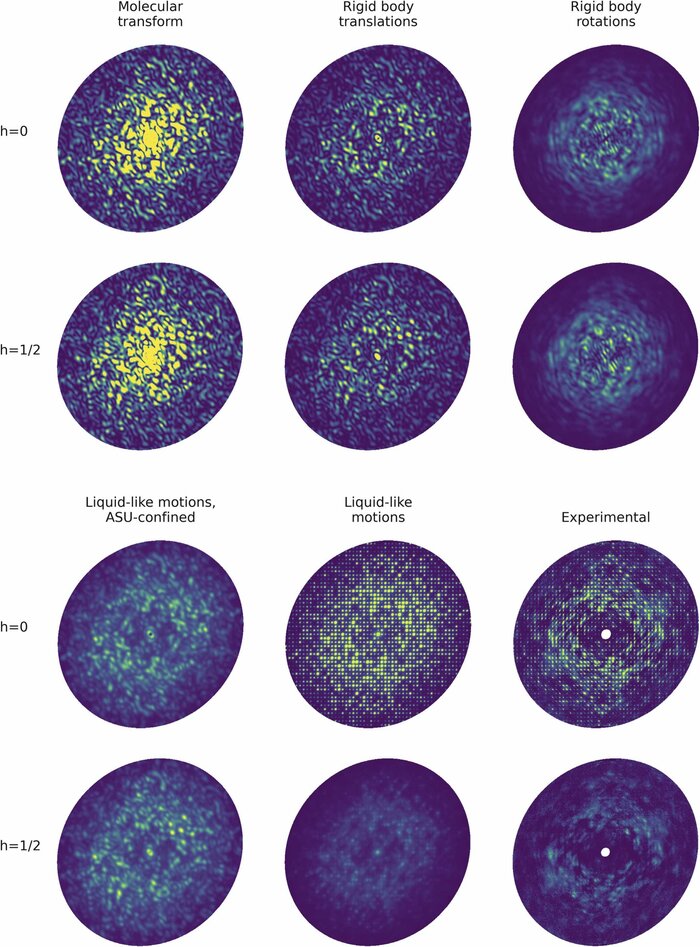 Modeling diffuse scattering with simple, physically interpretable modelsAriana Peck, Thomas J Lane, and Frédéric PoitevinIn Methods in enzymology, Nov 2023
Modeling diffuse scattering with simple, physically interpretable modelsAriana Peck, Thomas J Lane, and Frédéric PoitevinIn Methods in enzymology, Nov 2023Diffuse scattering has long been proposed to probe protein dynamics relevant for biological function, and more recently, as a tool to aid structure determination. Despite recent advances in measuring and modeling this signal, the field has not been able to routinely use experimental diffuse scattering for either application. A persistent challenge has been to devise models that are sophisticated enough to robustly reproduce experimental diffuse features but remain readily interpretable from the standpoint of structural biology. This chapter presents eryx, a suite of computational tools to evaluate the primary models of disorder that have been used to analyze protein diffuse scattering. By facilitating comparative modeling, eryx aims to provide insights into the physical origins of this signal and help identify the sources of disorder that are critical for reproducing experimental features. This framework also lays the groundwork for the development of more advanced models that integrate different types of disorder without loss of interpretability.





2022
- CryoEM
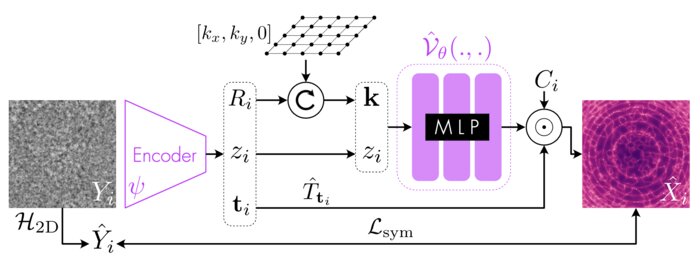 Amortized inference for heterogeneous reconstruction in cryo-emAxel Levy, Gordon Wetzstein, Julien NP Martel, and 2 more authorsAdvances in neural information processing systems, Nov 2022
Amortized inference for heterogeneous reconstruction in cryo-emAxel Levy, Gordon Wetzstein, Julien NP Martel, and 2 more authorsAdvances in neural information processing systems, Nov 2022Cryo-electron microscopy (cryo-EM) is an imaging modality that provides unique insights into the dynamics of proteins and other building blocks of life. The algorithmic challenge of jointly estimating the poses, 3D structure, and conformational heterogeneity of a biomolecule from millions of noisy and randomly oriented 2D projections in a computationally efficient manner, however, remains unsolved. Our method, cryoFIRE, performs ab initio heterogeneous reconstruction with unknown poses in an amortized framework, thereby avoiding the computationally expensive step of pose search while enabling the analysis of conformational heterogeneity. Poses and conformation are jointly estimated by an encoder while a physics-based decoder aggregates the images into an implicit neural representation of the conformational space. We show that our method can provide one order of magnitude speedup on datasets containing millions of images without any loss of accuracy. We validate that the joint estimation of poses and conformations can be amortized over the size of the dataset. For the first time, we prove that an amortized method can extract interpretable dynamic information from experimental datasets.
- CryoEM
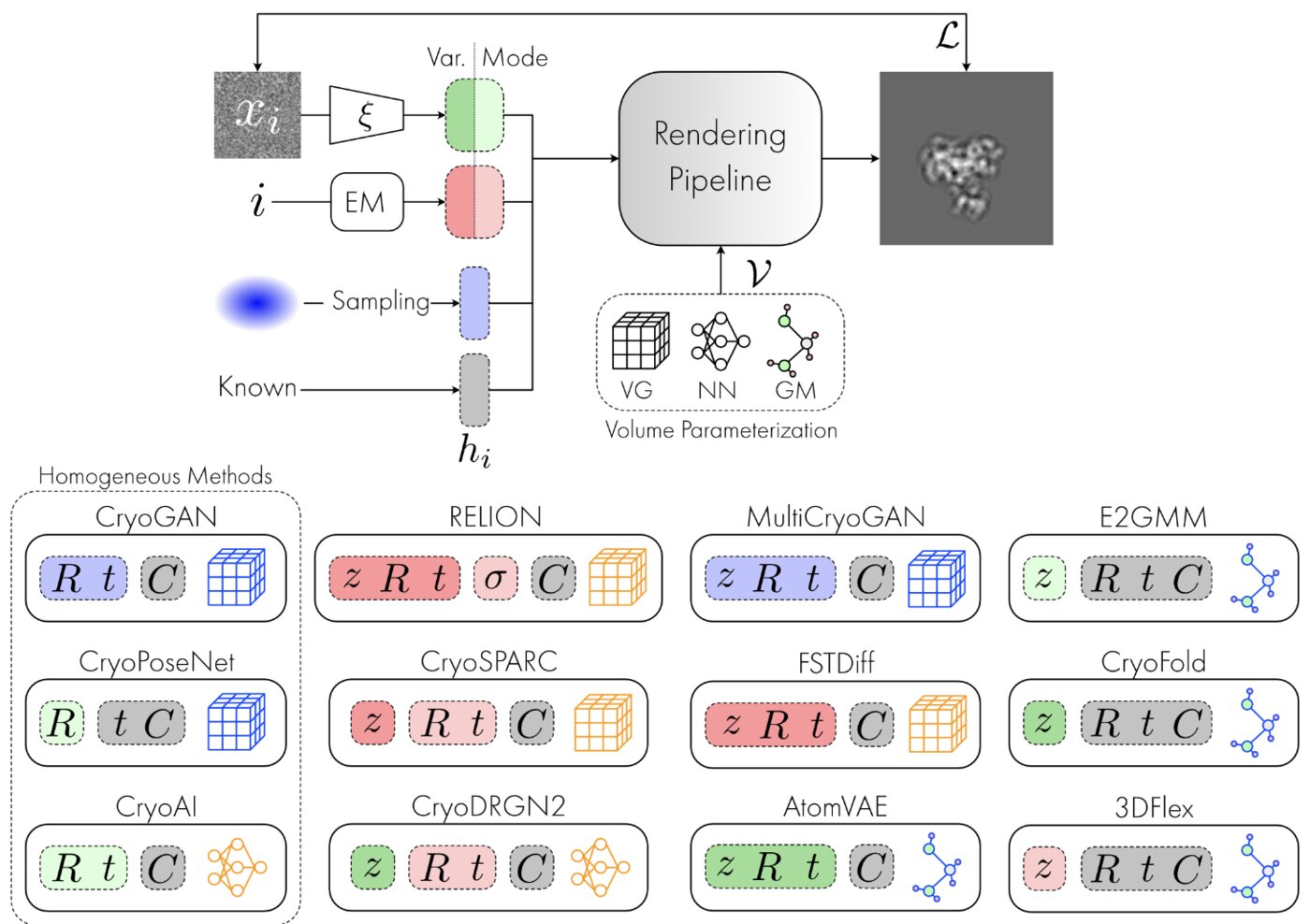 Deep generative modeling for volume reconstruction in cryo-electron microscopyClaire Donnat, Axel Levy, Frederic Poitevin, and 2 more authorsJournal of structural biology, Nov 2022
Deep generative modeling for volume reconstruction in cryo-electron microscopyClaire Donnat, Axel Levy, Frederic Poitevin, and 2 more authorsJournal of structural biology, Nov 2022Advances in cryo-electron microscopy (cryo-EM) for high-resolution imaging of biomolecules in solution have provided new challenges and opportunities for algorithm development for 3D reconstruction. Next-generation volume reconstruction algorithms that combine generative modelling with end-to-end unsupervised deep learning techniques have shown promise, but many technical and theoretical hurdles remain, especially when applied to experimental cryo-EM images. In light of the proliferation of such methods, we propose here a critical review of recent advances in the field of deep generative modelling for cryo-EM reconstruction. The present review aims to (i) provide a unified statistical framework using terminology familiar to machine learning researchers with no specific background in cryo-EM, (ii) review the current methods in this framework, and (iii) outline outstanding bottlenecks and avenues for improvements in the field.
- CryoEM
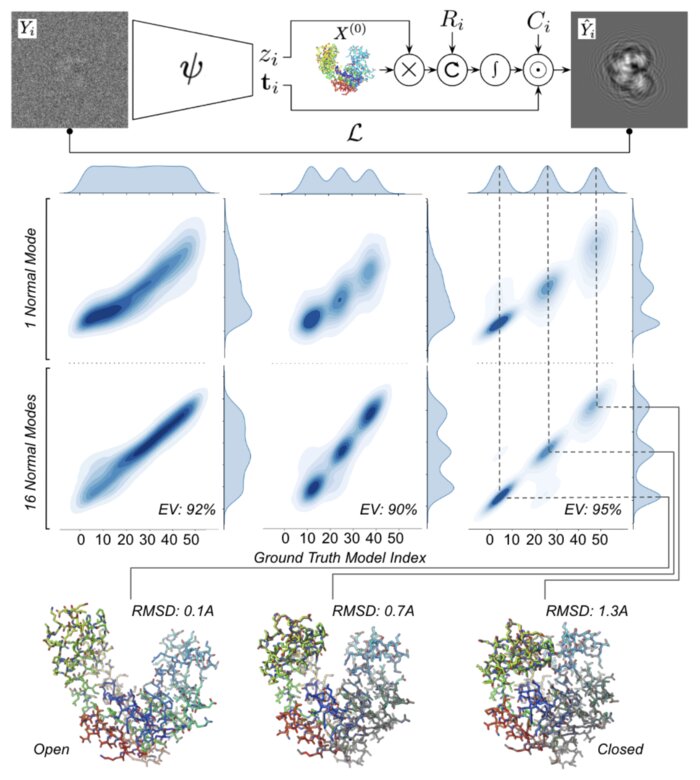 Heterogeneous reconstruction of deformable atomic models in Cryo-EMYoussef Nashed, Ariana Peck, Julien Martel, and 6 more authorsIn Machine learning for structural biology Workshop, Nov 2022
Heterogeneous reconstruction of deformable atomic models in Cryo-EMYoussef Nashed, Ariana Peck, Julien Martel, and 6 more authorsIn Machine learning for structural biology Workshop, Nov 2022Cryogenic electron microscopy (cryo-EM) provides a unique opportunity to study the structural heterogeneity of biomolecules. Being able to explain this heterogeneity with atomic models would help our understanding of their functional mechanisms but the size and ruggedness of the structural space (the space of atomic 3D cartesian coordinates) presents an immense challenge. Here, we describe a heterogeneous reconstruction method based on an atomistic representation whose deformation is reduced to a handful of collective motions through normal mode analysis. Our implementation uses an autoencoder. The encoder jointly estimates the amplitude of motion along the normal modes and the 2D shift between the center of the image and the center of the molecule . The physics-based decoder aggregates a representation of the heterogeneity readily interpretable at the atomic level. We illustrate our method on 3 synthetic datasets corresponding to different distributions along a simulated trajectory of adenylate kinase transitioning from its open to its closed structures. We show for each distribution that our approach is able to recapitulate the intermediate atomic models with atomic-level accuracy.
- CryoEM
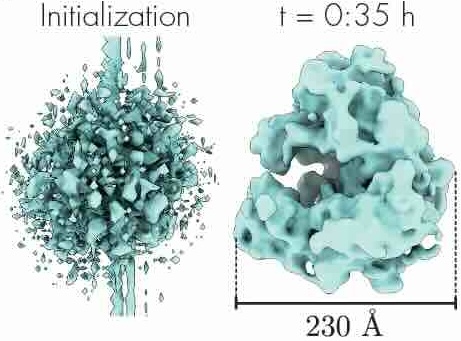 CryoAI: Amortized inference of poses for ab initio reconstruction of 3d molecular volumes from real cryo-em imagesAxel Levy, Frédéric Poitevin, Julien Martel, and 6 more authorsIn European Conference on Computer Vision, Nov 2022
CryoAI: Amortized inference of poses for ab initio reconstruction of 3d molecular volumes from real cryo-em imagesAxel Levy, Frédéric Poitevin, Julien Martel, and 6 more authorsIn European Conference on Computer Vision, Nov 2022Cryo-electron microscopy (cryo-EM) has become a tool of fundamental importance in structural biology, helping us understand the basic building blocks of life. The algorithmic challenge of cryo-EM is to jointly estimate the unknown 3D poses and the 3D electron scattering potential of a biomolecule from millions of extremely noisy 2D images. Existing reconstruction algorithms, however, cannot easily keep pace with the rapidly growing size of cryo-EM datasets due to their high computational and memory cost. We introduce cryoAI, an ab initio reconstruction algorithm for homogeneous conformations that uses direct gradient-based optimization of particle poses and the electron scattering potential from single-particle cryo-EM data. CryoAI combines a learned encoder that predicts the poses of each particle image with a physics-based decoder to aggregate each particle image into an implicit representation of the scattering potential volume. This volume is stored in the Fourier domain for computational efficiency and leverages a modern coordinate network architecture for memory efficiency. Combined with a symmetrized loss function, this framework achieves results of a quality on par with state-of-the-art cryo-EM solvers for both simulated and experimental data, one order of magnitude faster for large datasets and with significantly lower memory requirements than existing methods.
- Crystallography
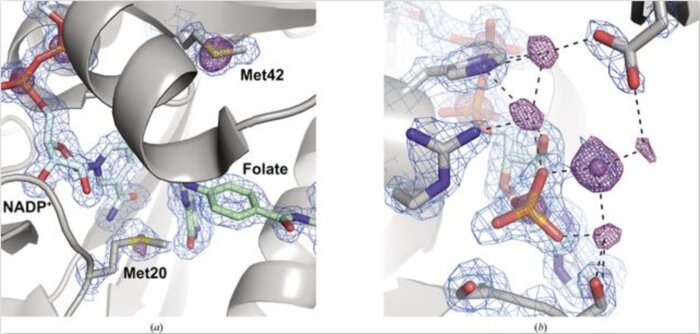 Native SAD phasing at room temperatureJ. B. Greisman, K. M. Dalton, C. J. Sheehan, and 3 more authorsNov 2022
Native SAD phasing at room temperatureJ. B. Greisman, K. M. Dalton, C. J. Sheehan, and 3 more authorsNov 2022Room-temperature crystallography enables researchers to resolve the conformational heterogeneity of structures. Here, the native SAD phasing of four structures at 295 K highlights the strengths of room-temperature diffraction experiments, including detailed anomalous difference maps and alternate conformations that are well supported by the electron density.
- Crystallography
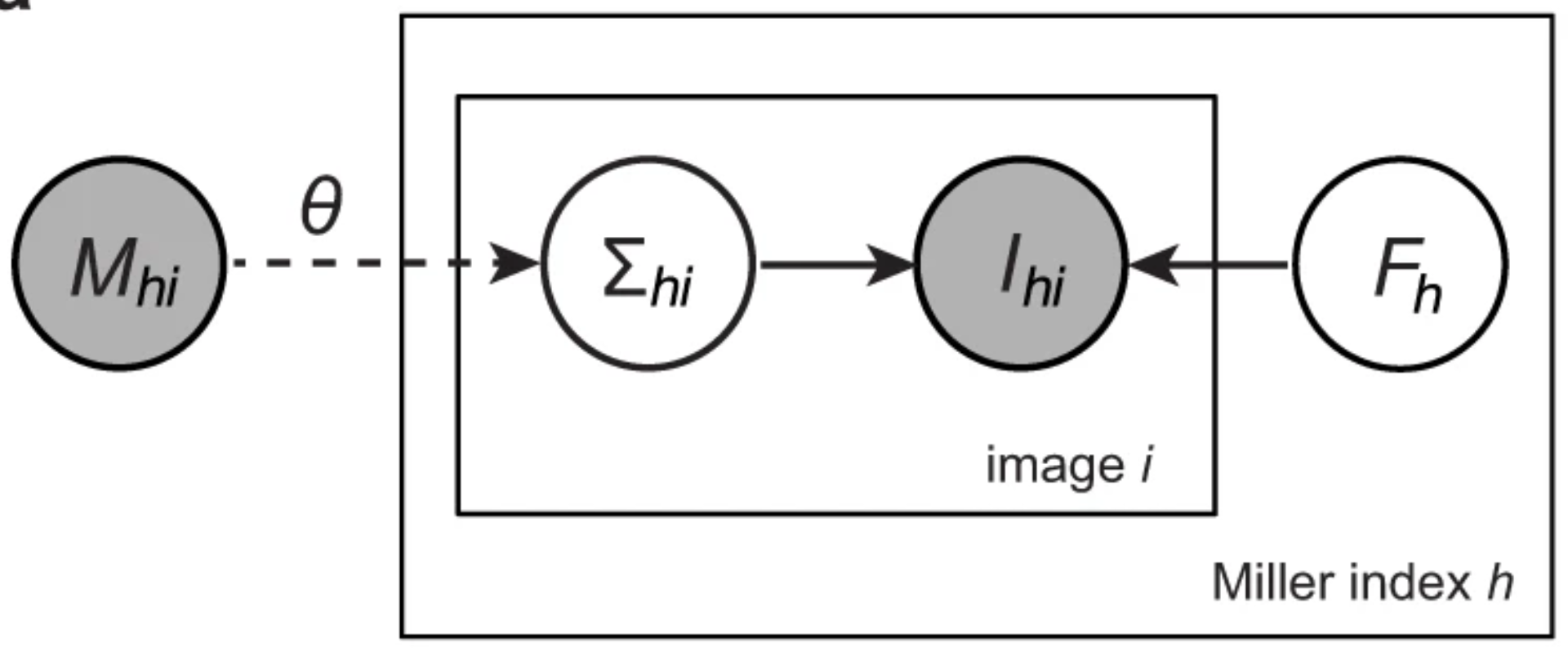 A unifying Bayesian framework for merging X-ray diffraction dataKevin M. Dalton, Jack B. Greisman, and Doeke R. HekstraNov 2022
A unifying Bayesian framework for merging X-ray diffraction dataKevin M. Dalton, Jack B. Greisman, and Doeke R. HekstraNov 2022Novel X-ray methods are transforming the study of the functional dynamics of biomolecules. Key to this revolution is detection of often subtle conformational changes from diffraction data. Diffraction data contain patterns of bright spots known as reflections. To compute the electron density of a molecule, the intensity of each reflection must be estimated, and redundant observations reduced to consensus intensities. Systematic effects, however, lead to the measurement of equivalent reflections on different scales, corrupting observation of changes in electron density. Here, we present a modern Bayesian solution to this problem, which uses deep learning and variational inference to simultaneously rescale and merge reflection observations. We successfully apply this method to monochromatic and polychromatic single-crystal diffraction data, as well as serial femtosecond crystallography data. We find that this approach is applicable to the analysis of many types of diffraction experiments, while accurately and sensitively detecting subtle dynamics and anomalous scattering.






2021
- Crystallography
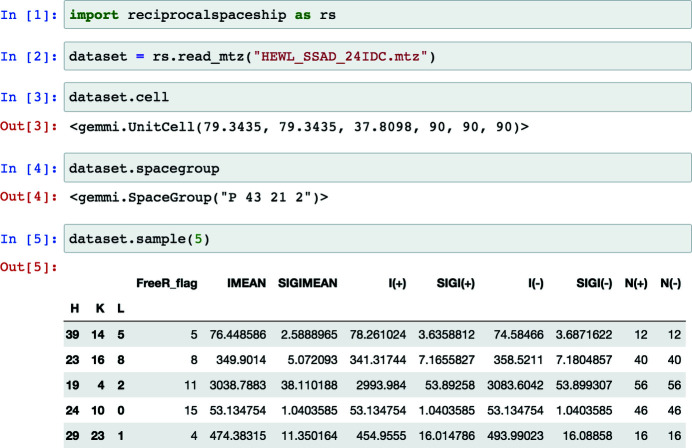 reciprocalspaceship: a Python library for crystallographic data analysisJack B. Greisman, Kevin M. Dalton, and Doeke R. HekstraNov 2021
reciprocalspaceship: a Python library for crystallographic data analysisJack B. Greisman, Kevin M. Dalton, and Doeke R. HekstraNov 2021Crystallography uses the diffraction of X-rays, electrons or neutrons by crystals to provide invaluable data on the atomic structure of matter, from single atoms to ribosomes. Much of crystallography’s success is due to the software packages developed to enable automated processing of diffraction data. However, the analysis of unconventional diffraction experiments can still pose significant challenges - many existing programs are closed source, sparsely documented, or challenging to integrate with modern libraries for scientific computing and machine learning. Described here is reciprocalspaceship, a Python library for exploring reciprocal space. It provides a tabular representation for reflection data from diffraction experiments that extends the widely used pandas library with built-in methods for handling space groups, unit cells and symmetry-based operations. As is illustrated, this library facilitates new modes of exploratory data analysis while supporting the prototyping, development and release of new methods.
- Diffuse Scattering
 Reproducibility of protein x-ray diffuse scattering and potential utility for modeling atomic displacement parametersZhen Su, Medhanjali Dasgupta, Frédéric Poitevin, and 5 more authorsStructural Dynamics, Nov 2021
Reproducibility of protein x-ray diffuse scattering and potential utility for modeling atomic displacement parametersZhen Su, Medhanjali Dasgupta, Frédéric Poitevin, and 5 more authorsStructural Dynamics, Nov 2021Protein structure and dynamics can be probed using x-ray crystallography. Whereas the Bragg peaks are only sensitive to the average unit-cell electron density, the signal between the Bragg peaks-diffuse scattering-is sensitive to spatial correlations in electron-density variations. Although diffuse scattering contains valuable information about protein dynamics, the diffuse signal is more difficult to isolate from the background compared to the Bragg signal, and the reproducibility of diffuse signal is not yet well understood. We present a systematic study of the reproducibility of diffuse scattering from isocyanide hydratase in three different protein forms. Both replicate diffuse datasets and datasets obtained from different mutants were similar in pairwise comparisons (Pearson correlation coefficient ≥0.8). The data were processed in a manner inspired by previously published methods using custom software with modular design, enabling us to perform an analysis of various data processing choices to determine how to obtain the highest quality data as assessed using unbiased measures of symmetry and reproducibility. The diffuse data were then used to characterize atomic mobility using a liquid-like motions (LLM) model. This characterization was able to discriminate between distinct anisotropic atomic displacement parameter (ADP) models arising from different anisotropic scaling choices that agreed comparably with the Bragg data. Our results emphasize the importance of data reproducibility as a model-free measure of diffuse data quality, illustrate the ability of LLM analysis of diffuse scattering to select among alternative ADP models, and offer insights into the design of successful diffuse scattering experiments.
- CryoEM
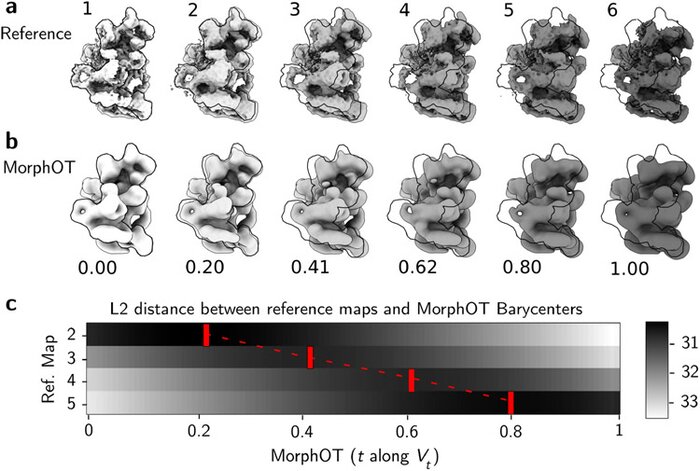 Application of transport-based metric for continuous interpolation between cryo-EM density mapsArthur Ecoffet, Geoffrey Woollard, Artem Kushner, and 2 more authorsAIMS mathematics, Nov 2021
Application of transport-based metric for continuous interpolation between cryo-EM density mapsArthur Ecoffet, Geoffrey Woollard, Artem Kushner, and 2 more authorsAIMS mathematics, Nov 2021Cryogenic electron microscopy (cryo-EM) has become widely used for the past few years in structural biology, to collect single images of macromolecules “frozen in time”. As this technique facilitates the identification of multiple conformational states adopted by the same molecule, a direct product of it is a set of 3D volumes, also called EM maps. To gain more insights on the possible mechanisms that govern transitions between different states, and hence the mode of action of a molecule, we recently introduced a bioinformatic tool that interpolates and generates morphing trajectories joining two given EM maps. This tool is based on recent advances made in optimal transport, that allow efficient evaluation of Wasserstein barycenters of 3D shapes. As the overall performance of the method depends on various key parameters, including the sensitivity of the regularization parameter, we performed various numerical experiments to demonstrate how MorphOT can be applied in different contexts and settings. Finally, we discuss current limitations and further potential connections between other optimal transport theories and the conformational heterogeneity problem inherent with cryo-EM data.
- CryoEM
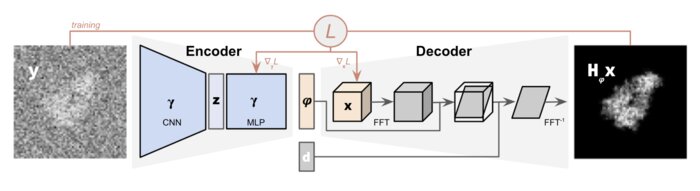 CryoPoseNet: End-to-end simultaneous learning of single-particle orientation and 3D map reconstruction from cryo-electron microscopy dataYoussef SG Nashed, Frédéric Poitevin, Harshit Gupta, and 4 more authorsIn Proceedings of the IEEE/CVF International Conference on Computer Vision, Nov 2021
CryoPoseNet: End-to-end simultaneous learning of single-particle orientation and 3D map reconstruction from cryo-electron microscopy dataYoussef SG Nashed, Frédéric Poitevin, Harshit Gupta, and 4 more authorsIn Proceedings of the IEEE/CVF International Conference on Computer Vision, Nov 2021Cryogenic electron microscopy (cryo-EM) provides images from different copies of the same biomolecule in arbitrary orientations. Here, we present an end-to-end unsupervised approach that learns individual particle orientations directly from cryo-EM data while reconstructing the 3D map of the biomolecule following random initialization. The approach relies on an auto-encoder architecture where the latent space is explicitly interpreted as orientations used by the decoder to form an image according to the physical projection model. We evaluate our method on simulated data and show that it is able to reconstruct 3D particle maps from noisy- and CTF-corrupted 2D projection images of unknown particle orientations.




2020
- CryoEM
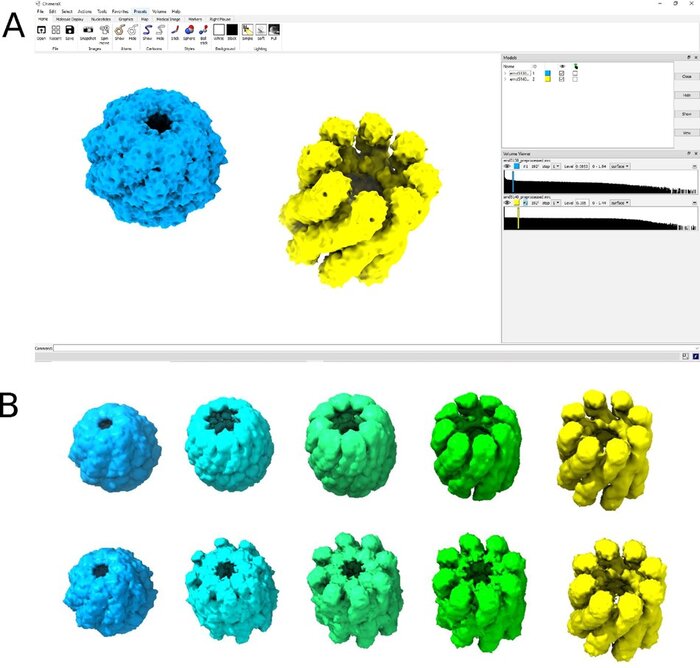 MorphOT: Transport-based interpolation between EM maps with UCSF ChimeraXArthur Ecoffet, Frédéric Poitevin, and Khanh Dao DucBioinformatics, Nov 2020
MorphOT: Transport-based interpolation between EM maps with UCSF ChimeraXArthur Ecoffet, Frédéric Poitevin, and Khanh Dao DucBioinformatics, Nov 2020Cryogenic electron microscopy (cryo-EM) offers the unique potential to capture conformational heterogeneity, by solving multiple three-dimensional classes that co-exist within a single cryo-EM image dataset. To investigate the extent and implications of such heterogeneity, we propose to use an optimal-transport-based metric to interpolate barycenters between EM maps and produce morphing trajectories.While standard linear interpolation mostly fails to produce realistic transitions, our method yields continuous trajectories that displace densities to morph one map into the other, instead of blending them.
- CryoEM
 Estimation of orientation and camera parameters from cryo-electron microscopy images with variational autoencoders and generative adversarial networksNina Miolane, Frédéric Poitevin, Yee-Ting Li, and 1 more authorIn Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition Workshops, Nov 2020
Estimation of orientation and camera parameters from cryo-electron microscopy images with variational autoencoders and generative adversarial networksNina Miolane, Frédéric Poitevin, Yee-Ting Li, and 1 more authorIn Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition Workshops, Nov 2020Cryo-electron microscopy (cryo-EM) is capable of producing reconstructed 3D images of biomolecules at near-atomic resolution. However, raw cryo-EM images are only highly corrupted - noisy and band-pass filtered - 2D projections of the target 3D biomolecules. Reconstructing the 3D molecular shape requires the estimation of the orientation of the biomolecule that has produced the given 2D image, and the estimation of camera parameters to correct for intensity defects. Current techniques performing these tasks are often computationally expensive, while the dataset sizes keep growing. There is a need for next-generation algorithms that preserve accuracy while improving speed and scalability. In this paper, we combine variational autoencoders (VAEs) and generative adversarial networks (GANs) to learn a low-dimensional latent representation of cryo-EM images. This analysis leads us to design an estimation method for orientation and camera parameters of single-particle cryo-EM images, which opens the door to faster cryo-EM biomolecule reconstruction.


2018
- Diffuse Scattering
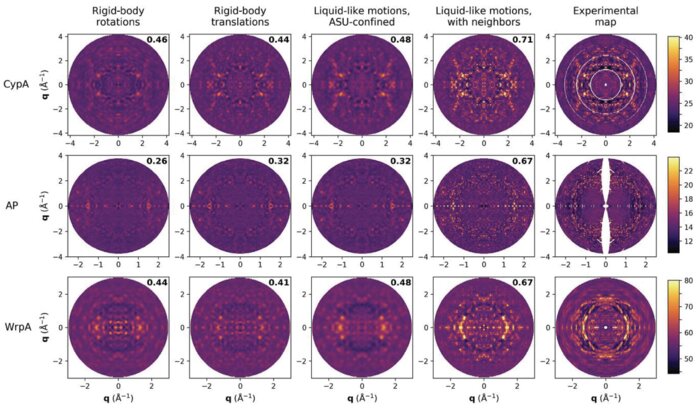 Intermolecular correlations are necessary to explain diffuse scattering from protein crystalsAriana Peck, Frédéric Poitevin, and Thomas J LaneIUCrJ, Nov 2018
Intermolecular correlations are necessary to explain diffuse scattering from protein crystalsAriana Peck, Frédéric Poitevin, and Thomas J LaneIUCrJ, Nov 2018Conformational changes drive protein function, including catalysis, allostery and signaling. X-ray diffuse scattering from protein crystals has frequently been cited as a probe of these correlated motions, with significant potential to advance our understanding of biological dynamics. However, recent work has challenged this prevailing view, suggesting instead that diffuse scattering primarily originates from rigid-body motions and could therefore be applied to improve structure determination. To investigate the nature of the disorder giving rise to diffuse scattering, and thus the potential applications of this signal, a diverse repertoire of disorder models was assessed for its ability to reproduce the diffuse signal reconstructed from three protein crystals. This comparison revealed that multiple models of intramolecular conformational dynamics, including ensemble models inferred from the Bragg data, could not explain the signal. Models of rigid-body or short-range liquid-like motions, in which dynamics are confined to the biological unit, showed modest agreement with the diffuse maps, but were unable to reproduce experimental features indicative of long-range correlations. Extending a model of liquid-like motions to include disorder across neighboring proteins in the crystal significantly improved agreement with all three systems and highlighted the contribution of intermolecular correlations to the observed signal. These findings anticipate a need to account for intermolecular disorder in order to advance the interpretation of diffuse scattering to either extract biological motions or aid structural inference.

2017
- Solution Scattering
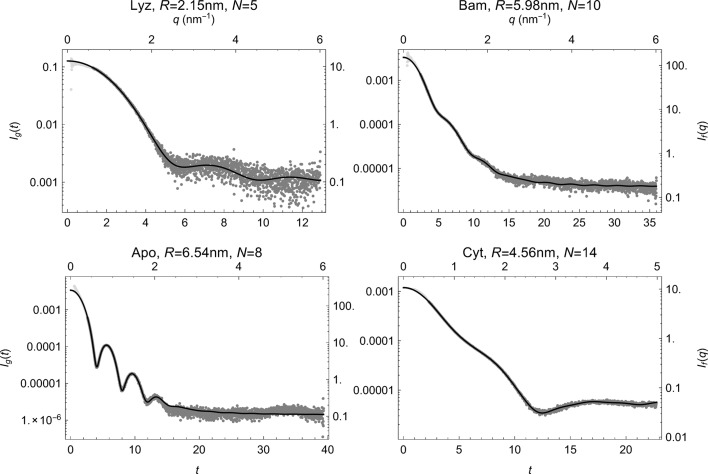 Reduction of small-angle scattering profiles to finite sets of structural invariantsJérome Houdayer, and Frédéric PoitevinActa Crystallographica Section A: Foundations and Advances, Nov 2017
Reduction of small-angle scattering profiles to finite sets of structural invariantsJérome Houdayer, and Frédéric PoitevinActa Crystallographica Section A: Foundations and Advances, Nov 2017It is shown how small-angle scattering (SAS) data can be reduced to a set of invariant parameters used to reliably estimate structural moments beyond the radius of gyration, thereby rigorously expanding the actual set of model-free quantities one can extract from experimental SAS data. The pair distance distribution function is also entirely described by this invariant set and the D max parameter can be measured.
