AtomicSPI
Learning atomic scale biomolecular dynamics from single-particle imaging data.
Frédéric Poitevin , Ellen Zhong , Gordon Wetzstein , Nina Miolane , Jay Shenoy , Axel Levy , David Klindt , Ariana Peck
Project Goal
The goal of the AtomicSPI project is to deliver software helping structural biologists resolve molecular conformations from single particle imaging datasets. The project work itself consists of four connected deliverables:
- An atomic representation of the particle: A graph-based atomic model will enable the application of physics-based priors during refinement, and will be fit directly to data, rather than fitting to an intermediate reconstructed 3D map (current practice).
- A deep learning model that maps individual particles to a continuous space of conformations and orientations: The deep learning model will capture the molecular dynamics crucial to understanding biological function in a feasible low-dimensional space. The model will provide a distribution of conformations (i.e. the energy landscape) with applications for establishing steady-state kinetic models.
- A differentiable digital twin of the electron microscope: The simulation will map the predicted structures and orientations to the images that would be produced by the electron microscope or X-ray FEL source. Crucially, by making the simulation differentiable, it can be inverted to infer structures directly from data, proposing new structures/orientations that correspond to an experimental image.
- A deep learning reconstruction pipeline: The pipeline will tie together the above three components to learn atomic models directly from measured datasets. By combining the three components into a single step, the proposed method will be both more efficient and more accurate than existing analysis pipelines.
Accomplishments
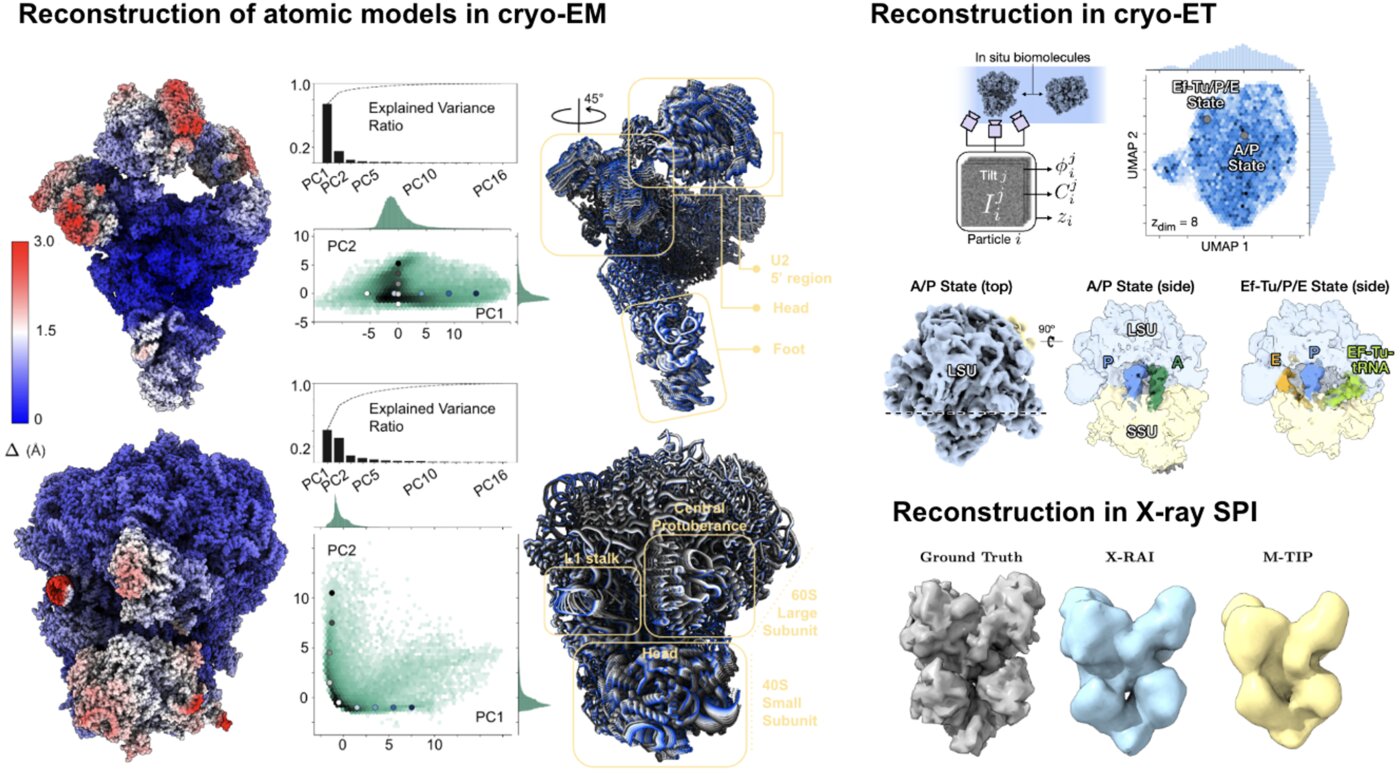
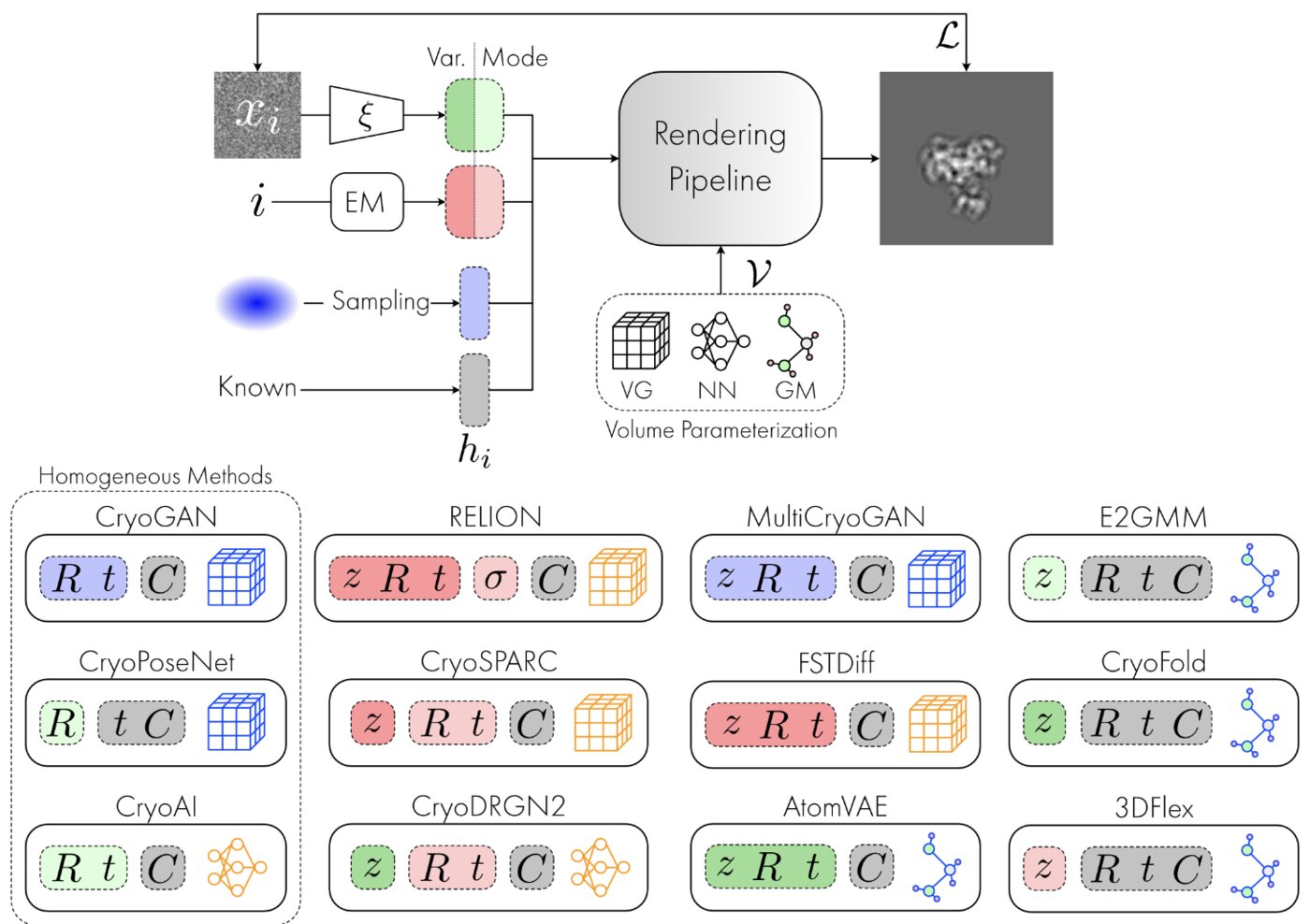
We have accomplished all four deliverables above in the cryoEM setting and prototyped them in the X-ray SPI setting. As summarized in Figure 1, the work carried thanks to this LDRD belongs to a new wave of next-generation volume reconstruction algorithm development that combines generative modeling with end-to-end unsupervised deep learning techniques.
We illustrate in Figure 2 our main achievements.
AtomicSPI Projects
For a deeper dive into the AtomicSPI projects, check out their individual pages:
Other directions explored in the project include studies on latent disentanglement of the conformational space and a general approach to solve inverse problems in protein space using diffusion-based priors.
Acknowledgements
This project sprung from discussions with Nina Miolane, following our initial work on cryo-EM image models. This project was supported by the LDRD program at SLAC from 2021 to 2024.
References
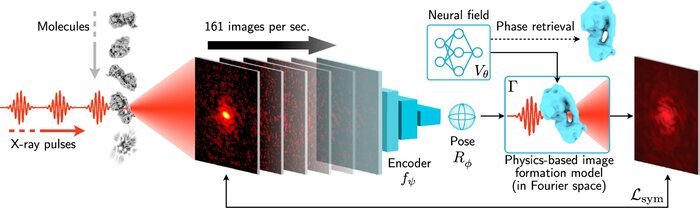
Scalable 3D reconstruction for X-ray single particle imaging with online machine learning
Nature Communications (2025)
X-ray free-electron lasers offer unique capabilities for measuring the structure and dynamics of biomolecules, helping us understand the basic building blocks of life. Notably, high-repetition-rate free-electron lasers enable single particle imaging, where individual, weakly scattering biomolecules are imaged under near-physiological conditions with the opportunity to access fleeting states that cannot be captured in cryogenic or crystallized conditions. Existing X-ray single particle reconstruction algorithms, which estimate the particle orientation for each image independently, are slow and memory-intensive when handling the massive datasets generated by emerging free-electron lasers. Here, we introduce X-RAI (X-Ray single particle imaging with Amortized Inference), an online reconstruction framework that estimates the structure of 3D macromolecules from large X-ray single particle datasets. X-RAI consists of a convolutional encoder, which amortizes pose estimation over large datasets, as well as a physics-based decoder, which employs an implicit neural representation to enable high-quality 3D reconstruction in an end-to-end, self-supervised manner. We demonstrate that X-RAI achieves state-of-the-art performance for small-scale datasets in simulation and challenging experimental settings and demonstrate its unprecedented ability to process large datasets containing millions of diffraction images in an online fashion. These abilities signify a paradigm shift in X-ray single particle imaging towards real-time reconstruction.
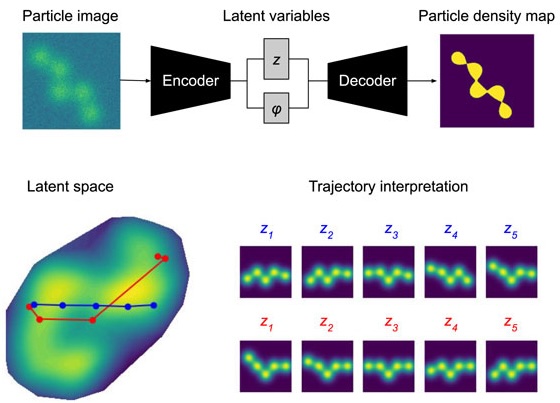
Towards interpretable Cryo-EM: disentangling latent spaces of molecular conformations
Frontiers in Molecular Biosciences (2024)
Molecules are essential building blocks of life and their different conformations (i.e., shapes) crucially determine the functional role that they play in living organisms. Cryogenic Electron Microscopy (cryo-EM) allows for acquisition of large image datasets of individual molecules. Recent advances in computational cryo-EM have made it possible to learn latent variable models of conformation landscapes. However, interpreting these latent spaces remains a challenge as their individual dimensions are often arbitrary. The key message of our work is that this interpretation challenge can be viewed as an Independent Component Analysis (ICA) problem where we seek models that have the property of identifiability. That means, they have an essentially unique solution, representing a conformational latent space that separates the different degrees of freedom a molecule is equipped with in nature. Thus, we aim to advance the computational field of cryo-EM beyond visualizations as we connect it with the theoretical framework of (nonlinear) ICA and discuss the need for identifiable models, improved metrics, and benchmarks. Moving forward, we propose future directions for enhancing the disentanglement of latent spaces in cryo-EM, refining evaluation metrics and exploring techniques that leverage physics-based decoders of biomolecular systems. Moreover, we discuss how future technological developments in time-resolved single particle imaging may enable the application of nonlinear ICA models that can discover the true conformation changes of molecules in nature. The pursuit of interpretable conformational latent spaces will empower researchers to unravel complex biological processes and facilitate targeted interventions. This has significant implications for drug discovery and structural biology more broadly. More generally, latent variable models are deployed widely across many scientific disciplines. Thus, the argument we present in this work has much broader applications in AI for science if we want to move from impressive nonlinear neural network models to mathematically grounded methods that can help us learn something new about nature.
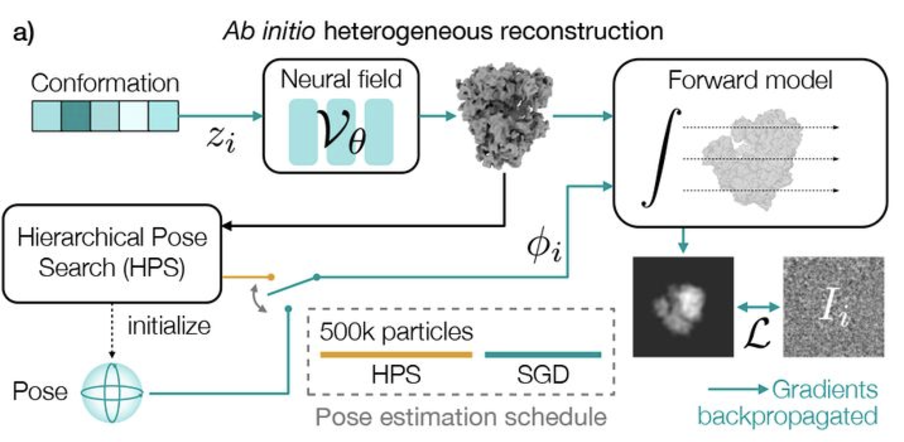
Revealing biomolecular structure and motion with neural ab initio cryo-EM reconstruction
bioRxiv (2024)
Proteins and other biomolecules form dynamic macromolecular machines that are tightly orchestrated to move, bind, and perform chemistry. Cryo-electron microscopy (cryo-EM) can access the intrinsic heterogeneity of these complexes and is therefore a key tool for understanding mechanism and function. However, 3D reconstruction of the resulting imaging data presents a challenging computational problem, especially without any starting information, a setting termed ab initio reconstruction. Here, we introduce a method, DRGN-AI, for ab initio heterogeneous cryo-EM reconstruction. With a two-step hybrid approach combining search and gradient-based optimization, DRGN-AI can reconstruct dynamic protein complexes from scratch without input poses or initial models. Using DRGN-AI, we reconstruct the compositional and conformational variability contained in a variety of benchmark datasets, process an unfiltered dataset of the DSL1/SNARE complex fully ab initio, and reveal a new “supercomplex” state of the human erythrocyte ankyrin-1 complex. With this expressive and scalable model for structure determination, we hope to unlock the full potential of cryo-EM as a high-throughput tool for structural biology and discovery.
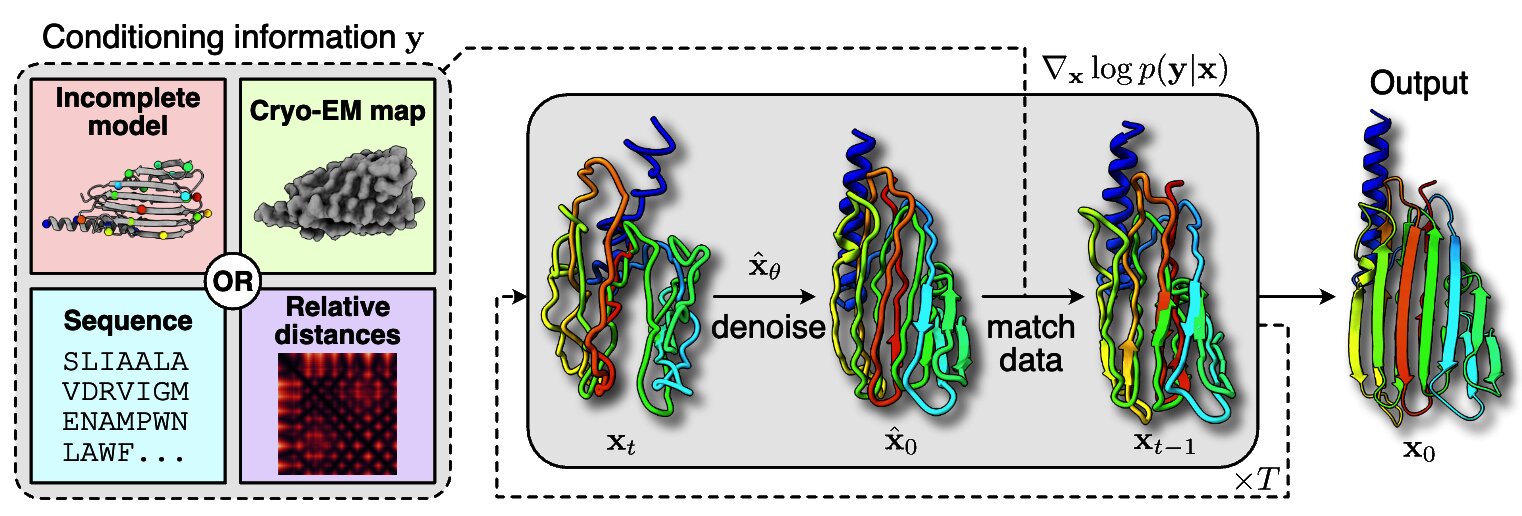
Solving Inverse Problems in Protein Space Using Diffusion-Based Priors
arXiv preprint arXiv:2406.04239 (2024)
The interaction of a protein with its environment can be understood and controlled via its 3D structure. Experimental methods for protein structure determination, such as X-ray crystallography or cryogenic electron microscopy, shed light on biological processes but introduce challenging inverse problems. Learning-based approaches have emerged as accurate and efficient methods to solve these inverse problems for 3D structure determination, but are specialized for a predefined type of measurement. Here, we introduce a versatile framework to turn raw biophysical measurements of varying types into 3D atomic models. Our method combines a physics-based forward model of the measurement process with a pretrained generative model providing a task-agnostic, data-driven prior. Our method outperforms posterior sampling baselines on both linear and non-linear inverse problems. In particular, it is the first diffusion-based method for refining atomic models from cryo-EM density maps.
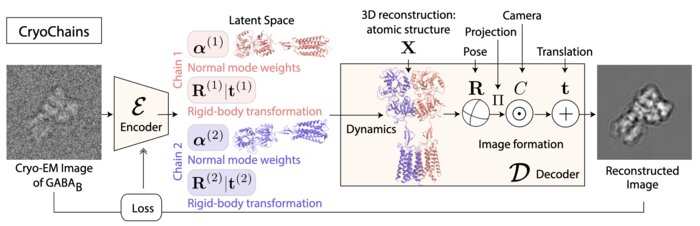
CryoChains: Heterogeneous Reconstruction of Molecular Assembly of Semi-flexible Chains from Cryo-EM Images
arXiv e-prints (2023)
Cryogenic electron microscopy (cryo-EM) has transformed structural biology by allowing to reconstruct 3D biomolecular structures up to nearatomic resolution. However, the 3D reconstruction process remains challenging, as the 3D structures may exhibit substantial shape variations, while the 2D image acquisition suffers from a low signal-to-noise ratio, requiring to acquire very large datasets that are time-consuming to process. Current reconstruction methods are precise but computationally expensive, or faster but lack a physically-plausible model of large molecular shape variations. To fill this gap, we propose CryoChains that encodes large deformations of biomolecules via rigid body transformation of their chains, while representing their finer shape variations with the normal mode analysis framework of biophysics. Our synthetic data experiments on the human GABAB and heat shock protein show that CryoChains gives a biophysicallygrounded quantification of the heterogeneous conformations of biomolecules, while reconstructing their 3D molecular structures at an improved resolution compared to the current fastest, interpretable deep learning method.
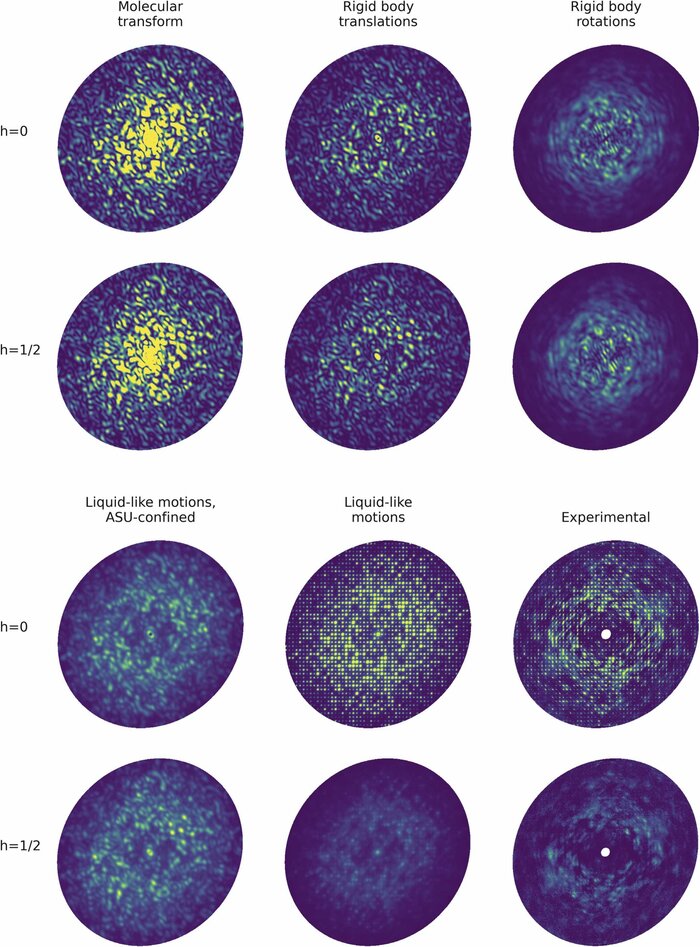
Modeling diffuse scattering with simple, physically interpretable models
Methods in enzymology (2023)
Diffuse scattering has long been proposed to probe protein dynamics relevant for biological function, and more recently, as a tool to aid structure determination. Despite recent advances in measuring and modeling this signal, the field has not been able to routinely use experimental diffuse scattering for either application. A persistent challenge has been to devise models that are sophisticated enough to robustly reproduce experimental diffuse features but remain readily interpretable from the standpoint of structural biology. This chapter presents eryx, a suite of computational tools to evaluate the primary models of disorder that have been used to analyze protein diffuse scattering. By facilitating comparative modeling, eryx aims to provide insights into the physical origins of this signal and help identify the sources of disorder that are critical for reproducing experimental features. This framework also lays the groundwork for the development of more advanced models that integrate different types of disorder without loss of interpretability.
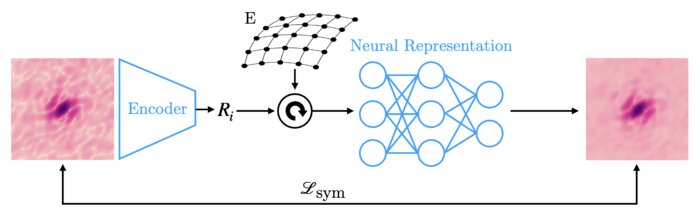
Amortized pose estimation for x-ray single particle imaging
Machine learning for structural biology Workshop (2023)
X-ray single particle imaging (SPI) is a nascent technique that can capture the dynamics of biomolecules at room temperature. SPI experiments will one day collect tens of millions of images of the same molecule in order to overcome the weak scattering of individual proteins. Existing reconstruction algorithms will be unable to scale to datasets of this size because they perform computationally expensive search steps to estimate the orientation of the molecule in each image. In this work, we propose a reconstruction algorithm that amortizes the estimation of pose via an autoencoder framework. Our approach consists of a convolutional encoder that maps X-ray images to predicted poses and a physics-based decoder that implicitly fuses all the 2D scattering images into a volumetric representation of the molecule. We validate our method on 6 synthetic datasets of 2 distinct proteins, showing that for the largest datasets containing 5 million images, our technique can reconstruct the electron density in a single pass.
Deep generative modeling for volume reconstruction in cryo-electron microscopy
Journal of structural biology (2022)
Advances in cryo-electron microscopy (cryo-EM) for high-resolution imaging of biomolecules in solution have provided new challenges and opportunities for algorithm development for 3D reconstruction. Next-generation volume reconstruction algorithms that combine generative modelling with end-to-end unsupervised deep learning techniques have shown promise, but many technical and theoretical hurdles remain, especially when applied to experimental cryo-EM images. In light of the proliferation of such methods, we propose here a critical review of recent advances in the field of deep generative modelling for cryo-EM reconstruction. The present review aims to (i) provide a unified statistical framework using terminology familiar to machine learning researchers with no specific background in cryo-EM, (ii) review the current methods in this framework, and (iii) outline outstanding bottlenecks and avenues for improvements in the field.
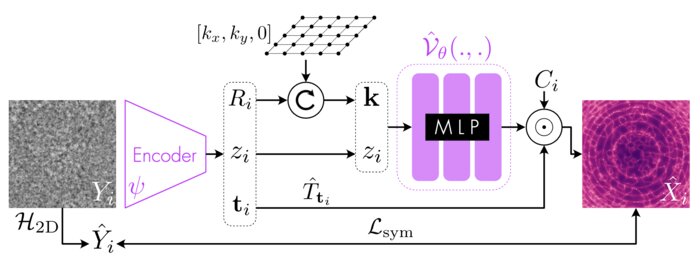
Amortized inference for heterogeneous reconstruction in cryo-em
Advances in neural information processing systems (2022)
Cryo-electron microscopy (cryo-EM) is an imaging modality that provides unique insights into the dynamics of proteins and other building blocks of life. The algorithmic challenge of jointly estimating the poses, 3D structure, and conformational heterogeneity of a biomolecule from millions of noisy and randomly oriented 2D projections in a computationally efficient manner, however, remains unsolved. Our method, cryoFIRE, performs ab initio heterogeneous reconstruction with unknown poses in an amortized framework, thereby avoiding the computationally expensive step of pose search while enabling the analysis of conformational heterogeneity. Poses and conformation are jointly estimated by an encoder while a physics-based decoder aggregates the images into an implicit neural representation of the conformational space. We show that our method can provide one order of magnitude speedup on datasets containing millions of images without any loss of accuracy. We validate that the joint estimation of poses and conformations can be amortized over the size of the dataset. For the first time, we prove that an amortized method can extract interpretable dynamic information from experimental datasets.
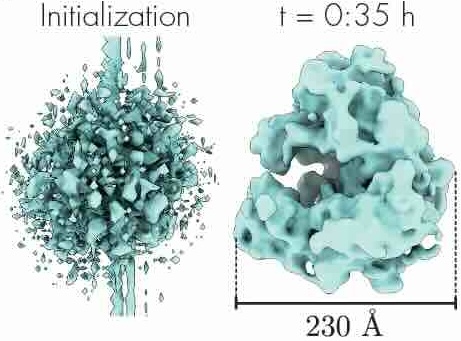
CryoAI: Amortized inference of poses for ab initio reconstruction of 3d molecular volumes from real cryo-em images
European Conference on Computer Vision (2022)
Cryo-electron microscopy (cryo-EM) has become a tool of fundamental importance in structural biology, helping us understand the basic building blocks of life. The algorithmic challenge of cryo-EM is to jointly estimate the unknown 3D poses and the 3D electron scattering potential of a biomolecule from millions of extremely noisy 2D images. Existing reconstruction algorithms, however, cannot easily keep pace with the rapidly growing size of cryo-EM datasets due to their high computational and memory cost. We introduce cryoAI, an ab initio reconstruction algorithm for homogeneous conformations that uses direct gradient-based optimization of particle poses and the electron scattering potential from single-particle cryo-EM data. CryoAI combines a learned encoder that predicts the poses of each particle image with a physics-based decoder to aggregate each particle image into an implicit representation of the scattering potential volume. This volume is stored in the Fourier domain for computational efficiency and leverages a modern coordinate network architecture for memory efficiency. Combined with a symmetrized loss function, this framework achieves results of a quality on par with state-of-the-art cryo-EM solvers for both simulated and experimental data, one order of magnitude faster for large datasets and with significantly lower memory requirements than existing methods.
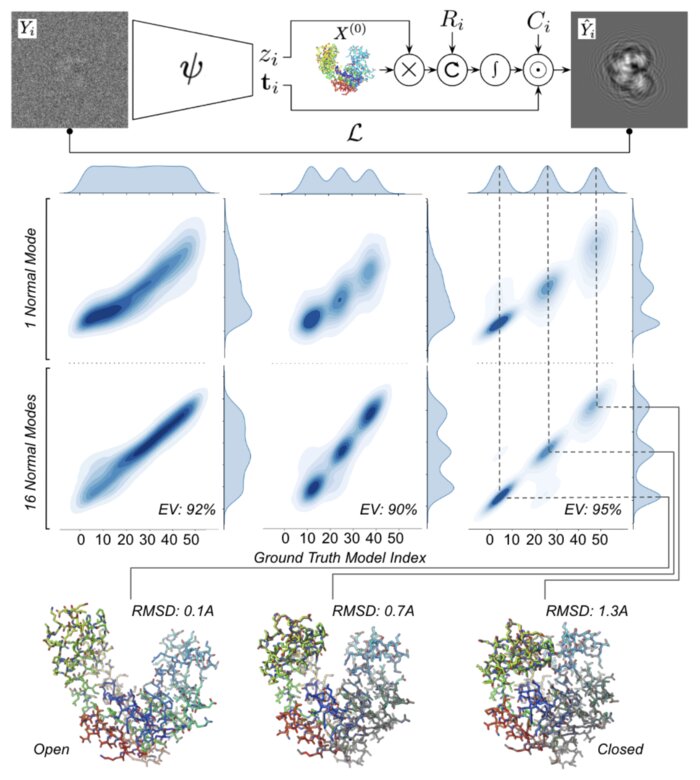
Heterogeneous reconstruction of deformable atomic models in Cryo-EM
Machine learning for structural biology Workshop (2022)
Cryogenic electron microscopy (cryo-EM) provides a unique opportunity to study the structural heterogeneity of biomolecules. Being able to explain this heterogeneity with atomic models would help our understanding of their functional mechanisms but the size and ruggedness of the structural space (the space of atomic 3D cartesian coordinates) presents an immense challenge. Here, we describe a heterogeneous reconstruction method based on an atomistic representation whose deformation is reduced to a handful of collective motions through normal mode analysis. Our implementation uses an autoencoder. The encoder jointly estimates the amplitude of motion along the normal modes and the 2D shift between the center of the image and the center of the molecule . The physics-based decoder aggregates a representation of the heterogeneity readily interpretable at the atomic level. We illustrate our method on 3 synthetic datasets corresponding to different distributions along a simulated trajectory of adenylate kinase transitioning from its open to its closed structures. We show for each distribution that our approach is able to recapitulate the intermediate atomic models with atomic-level accuracy.
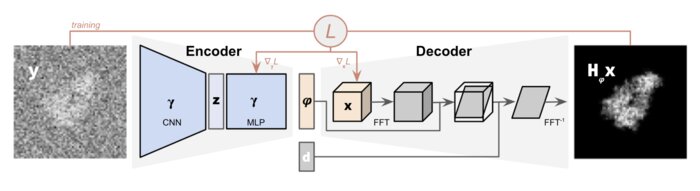
CryoPoseNet: End-to-end simultaneous learning of single-particle orientation and 3D map reconstruction from cryo-electron microscopy data
Proceedings of the IEEE/CVF International Conference on Computer Vision (2021)
Cryogenic electron microscopy (cryo-EM) provides images from different copies of the same biomolecule in arbitrary orientations. Here, we present an end-to-end unsupervised approach that learns individual particle orientations directly from cryo-EM data while reconstructing the 3D map of the biomolecule following random initialization. The approach relies on an auto-encoder architecture where the latent space is explicitly interpreted as orientations used by the decoder to form an image according to the physical projection model. We evaluate our method on simulated data and show that it is able to reconstruct 3D particle maps from noisy- and CTF-corrupted 2D projection images of unknown particle orientations.

Estimation of orientation and camera parameters from cryo-electron microscopy images with variational autoencoders and generative adversarial networks
Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition Workshops (2020)
Cryo-electron microscopy (cryo-EM) is capable of producing reconstructed 3D images of biomolecules at near-atomic resolution. However, raw cryo-EM images are only highly corrupted - noisy and band-pass filtered - 2D projections of the target 3D biomolecules. Reconstructing the 3D molecular shape requires the estimation of the orientation of the biomolecule that has produced the given 2D image, and the estimation of camera parameters to correct for intensity defects. Current techniques performing these tasks are often computationally expensive, while the dataset sizes keep growing. There is a need for next-generation algorithms that preserve accuracy while improving speed and scalability. In this paper, we combine variational autoencoders (VAEs) and generative adversarial networks (GANs) to learn a low-dimensional latent representation of cryo-EM images. This analysis leads us to design an estimation method for orientation and camera parameters of single-particle cryo-EM images, which opens the door to faster cryo-EM biomolecule reconstruction.